Given our patient’s clinical history and exam, a differential diagnosis was constructed and included conditions that were associated with optic neuritis. Notably, we were concerned about vascular (giant cell arteritis, nonarteritic anterior ischemic optic neuropathy); demyelinating (multiple sclerosis); autoimmune (systemic lupus erythematous, Sjögren’s syndrome); infectious (tuberculosis, herpes zoster, Lyme disease, syphilis, toxoplasmosis, Bartonella); and inflammatory (sarcoidosis) etiologies. Given the clinical presentation, suspicion for giant cell arteritis was not high. The presence of a crowded optic disc in the left eye along with hypertension and older age was highly concerning for nonarteritic anterior ischemic optic neuropathy (NAION).
Humphrey visual field testing was reliable and confirmed the inferior nasal defect in her right eye and was otherwise normal in the left eye. Optical coherence tomography demonstrated retinal nerve fiber layer thickness of 123 μm in the right eye with significant swelling and 85 μm in the left with atrophy.
Magnetic resonance imaging of the brain demonstrated several nonspecific scattered punctate foci of FLAIR hyperintense signals within the cerebral white matter, predominantly in a subcortical distribution. These signs were suspicious for small vessel ischemic changes. Additionally, there were several larger oval lesions suspicious for demyelinating disease, but not specific for MS (See Figure 2). There was no evidence of acute ischemia, intracranial hemorrhage or masses. MRI examination of the orbits was unremarkable and did not disclose optic nerve enhancement. Magnetic resonance angiography identified an incidental medially directed 6-mm left paraclinoid ICA aneurysm and 2-mm aneurysm from right ACA. CT of the chest was within normal limits and did not demonstrate evidence of tuberculosis.
Laboratory evaluation was within normal limits except for Neuromyelitis Optica (NMO) antibody against aquaporin-4 (AQP4) at a positive value of 6.3 (>5), which is consistent with diagnosis of or predisposition to NMO.
Following the diagnosis of NMO or Devic’s disease, the patient was started on 50 mg of prednisone and instructed to taper down by 10 mg every five days until she returned to her original dosage of 5 mg, as per her rheumatologist for her RA. In conjunction with her rheumatologist, she was restarted on rituximab infusions.
Discussion
NMO or Devic’s disease is an autoimmune disorder that results from inflammation and demyelination of the optic nerves and spinal cord. Originally identified in 1894 by Dr. Eugène Devic, NMO can result in blindness, weakness/paraplegia, loss of sensation, bowel and bladder dysfunction as well as other symptoms.1 In this regard, NMO shares some similarities with MS and was initially proposed to be on the MS spectrum. However, recent immunological, clinical, radiographical and pathological studies delineate NMO as a unique disease. The prevalence of NMO is believed to be between 0.5 and 4.4 per 100,000 individuals and does not appear to have racial predilection.2,3 The average age of onset for NMO is 34 to 43 years with a range of 4 to 88 years old.2,4 Numerous studies have documented that women are three- to tenfold more likely to be diagnosed with NMO as compared to men.2,5
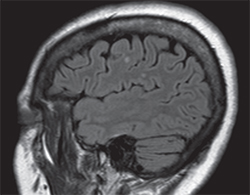 |
| Figure 2. Sagittal magnetic resonance imaging demonstrating FLAIR hyperintense signals in cerebral white matter. |
MS is roughly 40 times more prevalent than NMO.4 Distinguishing NMO from MS is important clinically due to the fact that MS treatments of IFN-β, natalizumab and oral fingolimod may exacerbate NMO in certain patients.4,10 The recently updated diagnostic criteria for NMO with positive AQP4 antibody requires at least one core clinical characteristic (optic neuritis or transverse myelitis) and exclusion of alternative diagnoses (notably brain MRI not meeting usual diagnostic criteria for MS).8
Treatment options for NMO are currently based on small, mostly retrospective studies—to date there have been no prospective randomized clinical trials to establish efficacy of current treatments. With acute presentation, methylprednisolone 1g IV is delivered daily for three to five days. If symptoms do not improve, plasma exchange is administered daily or every other day for up to five treatments.4 Afterwards, systemic immunosuppression with a variety of agents (including azathioprine, mycophenolate, cyclophosphamide and rituximab) have been employed to prevent attack recurrences.4,10 Several studies have demonstrated rituximab treatment performs superiorly at decreasing annual relapse rates and disability indices.11,12
As compared to MS, the myelopathy in NMO is typically more severe and has less of a chance of recovery.10 Additionally, the transverse myelitis in the spine can cause respiratory failure and death.13 Within the first year, 55 percent of patients will have a relapse, and 90 percent of patients will do so in the first five years—these neurological deficits usually do not resolve between episodes.2
In summary, NMO is an autoimmune disease resulting in demyelination and inflammation of the optic nerves and spinal cord from autoantibodies directed against aquaporin-4. NMO may manifest with loss of vision or spinal-cord dysfunction and may be clinically similar to MS. Diagnosis is based primarily on MRI, evidence of optic neuritis and transverse myelitis, presence of IgG against AQP4, and exclusion of alternative diagnoses. Acutely, patients are treated with steroid or plasmapheresis before using systemic immunosuppression to prevent recurrent attacks. Without treatment, prognosis for NMO is guarded. REVIEW
1. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004;364(9451):2106-2112.
2. Pereira WL, Reiche EM, Kallaur AP, Kaimen-Maciel DR. Epidemiological, clinical, and immunological characteristics of neuromyelitis optica: A review. J Neurol Sci 2015;355(1-2):7-17.
3. Pandit L, Asgari N, Apiwattanakul M, et al. Demographic and clinical features of neuromyelitis optica: A review. Mult Scler 2015;21(7):845-853.
4. Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: State-of-the-art and emerging therapies. Nat Rev Neurol 2014;10(9):493-506.
5. Collongues N, Marignier R, Zéphir H, et al. Neuromyelitis optica in France: A multicenter study of 125 patients. Neurology 2010;74(9):736-742.
6. Jarius S, Franciotta D, Bergamaschi R, et al. NMO-IgG in the diagnosis of neuromyelitis optica. Neurology 2007;68(13):1076-1077.
7. Jarius S, Paul F, Franciotta D, et al. Mechanisms of disease: Aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 2008;4(4):202-214.
8. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85(2):177-189.
9. Ruiz-Gaviria R, Baracaldo I, Castañeda C, Ruiz-Patiño A, Acosta-Hernandez A, Rosselli D. Specificity and sensitivity of aquaporin 4 antibody detection tests in patients with neuromyelitis optica: A meta-analysis. Mult Scler Relat Disord 2015;4(4):345-349.
10. Cabre P, Olindo S, Marignier R, et al. Efficacy of mitoxantrone in neuromyelitis optica spectrum: Clinical and neuroradiological study. J Neurol Neurosurg Psychiatry 2013;84(5):511-516.
11. Torres J, Pruitt A, Balcer L, Galetta S, Markowitz C, Dahodwala N. Analysis of the treatment of neuromyelitis optica. J Neurol Sci 2015;351(1-2):31-35.
12. Kim SH, Jeong IH, Hyun JW, et al. Treatment Outcomes With Rituximab in 100 Patients With Neuromyelitis Optica: Influence of FCGR3A Polymorphisms on the Therapeutic Response to Rituximab. JAMA Neurol 2015 Sep 1;72(9):989-95.
13. Hoorbakht H, Bagherkashi F. Optic neuritis, its differential diagnosis and management. Open Ophthalmol J 2012;6:65-72.


