Given our patient’s clinical history and exam, a differential diagnosis was constructed and included conditions that could cause pigmentary changes and scotomas. This included inflammatory causes such as sarcoidosis; multiple evanescent white dot syndrome; acute zonal occult outer retinopathy; acute retinal pigment epitheliitis; and unilateral idiopathic maculopathy. Additional conditions that could produce unilateral paracentral scotomas were included, such as acute macular neuroretinopathy and paracentral acute middle maculopathy. The differential also included hereditary conditions such as Stargardt’s disease; cone dystrophy; and cone-rod dystrophy, along with infectious causes such as histoplasmosis, syphilis and tuberculosis.
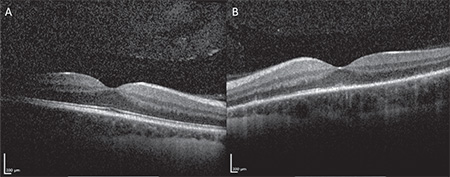
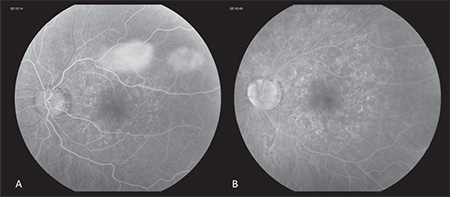
The patient was reevaluated five days later, at which time her visual acuity was 20/20 OD and 20/100 OS. External, anterior and posterior exam were unchanged from prior. At this time, spectral-domain optical coherence tomography was performed OU (See Figure 3a, b), and although unremarkable OD, clearly highlighted outer retinal deposits OS and significant disruption of the ellipsoid layer. Fluorescein angiography (See Figure 4a, b) OS demonstrated wreath-like early and late hyperfluorescence corresponding to the pigmentary changes noted on clinical exam. At this point, the diagnosis of multiple evanescent white dot syndome was made. The patient was observed over the next month. Upon follow-up one month later, her visual acuity improved to 20/20 OD and 20/25 OS, and fundus examination demonstrated resolution of the pigmentary changes OS.
|
Discussion
Multiple evanescent white dot syndrome (MEWDS) was first described by Lee M. Jampol, MD, and colleagues in 1984.1 It makes up one of the White Dot Syndome disorders, along with acute posterior multifocal placoid pigment epitheliopathy (APMPPE); birdshot chorioretinopathy; diffuse unilateral subacute retinitis; multifocal choroiditis with panuveitis (MCP); serpiginous choroiditis; and acute zonal occult outer retinopathy (AZOOR). The White Dot Syndrome disorders are differentiated according to clinical history, classic examination findings, laterality of pathology and ancillary testing including fluorescein angiography and fundus autofluorescence.
MEWDS typically affects young patients between the ages of 20 and 45, and has a predilection for females. Findings are generally unilateral, although bilateral cases have been reported.2 Symptoms of MEWDS include temporal or paracentral scotomas, dyschromatopsia, photopsias and blurred vision, although visual acuity can range from 20/20 to 20/400.3,4
Fundus examination can demonstrate mild amounts of vitreous cell, hyperemia or edema of the optic disc and retinal sheathing. Classically, small white dots are noticed in the outer layers of the retina and retinal pigment epithelium, which are often subtle and self-resolving. Visual field testing often demonstrates scotomas that correlate with retinal findings or an enlarged blind spot, and fluorescein angiography demonstrates early punctate hyperfluorescence in a wreath-like pattern and late staining of the white dots.3,4 Fundus autofluorescence has been proven to be a valuable tool in highlighting RPE changes, even in the absence of clear white dots on fundus examination.5 The exact pathophysiology of the pigmentary alterations remains unknown, although recent reports speculate that photoreceptor loss, as demonstrated on SD-OCT, may unmask the underlying RPE autofluorescence.5,6
The cause of MEWDS is not entirely clear, although it is theorized to be preceded by a viral illness such as influenza in the majority of cases. It has also been reported in association with influenza, hepatitis B and hepatitis C vaccinations.7,8,9 The clinical course of MEWDS is self-limited and resolves in a majority of cases without any need for treatment. However, scotomata, photopsias and dyschromotopsia may persist.3,4 REVIEW
1. Jampol LM, Sieving PA, Pugh D, et al. Multiple evanescent white dot syndrome. Arch Ophthalmol 1984;102:671-4.
2. Tsai L, Jampol LM, Pollock SC, et al. Chronic recurrent multiple evanescent white dot syndrome. Retina 1994;14:160-63.
3. Quillen DA, Davis JB, Gottlieb JL, et al. The white dot syndromes. Am J Ophthalmol 2004;137:538-50.
4. Jampol LM, Wiredu A. MEWDS, MFC, PIC, AMN, AIBSE, and AZOOR: One disease or many? Retina 1995;15:373-378.
5. Joseph A, Rahimy E, Freund KB, et al. Fundus autofluoresence and photoreceptor bleaching in multiple evanescent white dot syndrome. Ophthalmic Surg Lasers Imaging Retina 2013;44(6):588-92.
6. Thomas BJ, Albini TA, Flynn HW. Multiple evanescent white dot syndrome: Multimodal imaging and correlated with proposed pathophysiology. Ophthalmic Surg Lasers Imaging Retina 2013;44:584-7.
7. Goyal S, Nazarian SM, Thayi DR, et al. Multiple evanescent white dot syndrome following recent influenza vaccination. Can J Ophthalmol 2013;48(5):115-6.
8. Fine L, Fine A, Cunningham ET Jr. Multiple evanescent white dot syndrome following hepatitis A vaccination. Arch Ophthalmol 2001;119:1856-8.
9. Baglivo E, Safran AB, Borruat FX. Multiple evanescent white dot syndrome after hepatitis B vaccine. Am J Ophthalmol 1996;122:431-2.


