It’s not an unusual situation for most practitioners: In your exam room sits a new patient, presenting with a host of signs and symptoms that define a specific disorder, disease X. After a thorough history, physical examination, and appropriate clinical testing, you, as the knowledgeable physician, prescribe the drug therapy sin qua non for disease X. Two weeks later the patient is back in your office, complaining that the therapy did not work. What went wrong? If we assume that patient compliance (a common culprit) is not an issue, then the next most likely explanation is that it’s the patient’s fault!
In this month’s column we’ll discuss the topic of therapeutic non-responders and, in the process, touch on noteworthy aspects of drug development, drug metabolism and the genetic underpinnings of some of the examples of therapeutic non-responders we’re familiar with, and some new ones as well.

Why blame the patient? Of course, treatment response rates for specific therapeutic regimens are known to vary based on age, gender, disease severity, ethnicity, concomitant medication use and other factors. And environmental variables can certainly play a role as well. These are issues that are typically taken into account at the time of diagnosis, and so for them the burden is on the practitioner. But the most recent studies are revealing a far greater impact of genetics in determining the nature of drug responses—and your patients brought their unresponsive genes with them when they walked into your office.
With these developments in mind, how can we improve on rates of treatment response, and how can we develop better methods to predict which treatment approach will work best for each patient?
Before we consider the basis of “personalized medicine” we’ll start with examples of response variability, and then briefly discuss what part the therapy itself might play.
The Classic Non-Responder
One of the best examples of variable response in therapy for ophthalmic disorders is in dry-eye disease. The only currently available prescription product for treatment of dry eye is cyclosporine 0.05% emulsion (Restasis), but this product is efficacious only for a fraction (around 15 percent) of all dry-eye patients.1 While that figure might seem like it results in an 85-percent non-responder rate, in fact the label indication for this drug states that it is specifically intended for only a subset of all dry-eye patients. This is an example that highlights the complexities of the phenomenon of non-responders. In this case, low apparent response rates among all dry-eye patients are due to the heterogeneity of the disease itself, which, despite the overall designation of “dry-eye disease” is actually a collection of different conditions (e.g., aqueous tear deficiency, meibomian gland dysfunction, etc.), each with distinct etiologies and a shared spectrum of symptoms reported by patients.2
In other disorders, including glaucoma, age-related macular degeneration and ocular allergies, there is a similar spectrum of etiologies and a corresponding set of standard therapies. And for all of these there are some patients who don’t respond in a clinically meaningful way. In some of these cases, the lack of response is more about the drug that’s been prescribed than the patient.
Non-Responders in Trials
An aspect of non-responders that is often overlooked is the role of the drug development process. It’s worth noting that to get a drug approved by the Food and Drug Administration, clinical study results aren’t required to show which population characteristics dictate the likelihood of positive drug response. With this being the case, pharmaceutical companies will simply select subjects who have the disease and who, as much as possible, are otherwise representative of the affected population.
Clinical trials also impact the level of non-responders to a new therapy by virtue of the measures of drug efficacy selected. The selection of an endpoint or endpoints in a clinical trial, while designed to provide reliable metrics for drug efficacy, can create inclusive or exclusive subgroups within a larger population. If the endpoint is based upon a statistical target that is population-based, a “super-responder” subpopulation can pull less responsive sub-populations into the range of statistical significance.
An approach to avoid such occurrences is the use of a responder analysis, in which subjects are dichotomized into responders or non-responders.3 The goal of this analysis is to distinguish statistical significance from clinical significance, and thereby minimize the likelihood that a therapy will be ineffective in the general population. Unfortunately, this method is limited by both a lack of statistical power (and it has significant cost issues as a result) and by the inherently arbitrary designation of cutoff points between responder and non-responder groups.
Despite these issues, responder analysis can be a useful approach under the appropriate circumstances. For example, a 1997 study of dolasetron4 used “complete response” as an endpoint for prevention of postoperative nausea and vomiting following surgery. Complete response was defined as the absence of emetic episodes and no usage of escape medication during the 24 hours following surgery. The authors reported that a higher proportion of complete response was observed in the treatment group vs. the placebo group. This example demonstrates that for some studies the designation of responder is unambiguous.
Other studies take the issue of non-responders head-on, enrolling patients with a history of non-response to an available therapy. One study investigated the use of daily consensus interferon and ribavirin therapy in a population of patients who demonstrated non-response to previous IFN or IFN/ribavirin therapy.5 While the study demonstrated some improvement in virological response, a high relapse rate was observed after treatment cessation. Furthermore, the authors discuss several substantial challenges to non-responder studies, including the potential for enrolling a heterogeneous group of non-responders with a variety of reasons for non-response and the ongoing necessity of meticulously monitoring compliance to be sure non-response isn’t actually a result of non-compliance. This last point brings us back to the initial premise that when therapy fails, it’s not unreasonable to blame the patient. But for those who follow their doctor’s orders, the explanation for lack of response to therapy seems to be turning into an issue of genetics.
 Pharmacogenetics
Pharmacogenetics
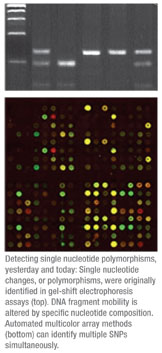
Hope for many of those in the non-responder populations lies, at least in part, in our ability to understand the role human diversity plays in the effectiveness of the drugs we develop. In the pre-human-genome era, the conventional wisdom was that genetic variation in patients was unlikely to play a significant role in the observed variability in drug responses. This was based on an expectation that genetic variation itself was uncommon. Now, however, we know that this is not the case: On average, comparison of genomes of two unrelated individuals will reveal approximately 3 million SNPs, or single nucleotide polymorphisms, between them. Each of these polymorphisms represents a single change in their respective genetic codes, and collectively represents about 1 percent of the human genetic code. While many of these changes are silent—in other words they don’t alter the gene product—most recent evidence suggests that up to half of the inter-individual differences in drug response may be related to genetic variability. This area of medicine is described by pharmacogenomics, the field of study dedicated to investigating genetic variations and response to therapeutics; and pharmacogenetics, the area of research that examines mutations in individual genes responsible for drug metabolism and drug action.
In a general sense, there are two pathways through which genetic variation can alter a patient’s response to a specific drug. Atypical responses can be due to individual genetic variations leading to altered activity of the enzyme or enzymes responsible for metabolism of the drug. This type of genetic variation most commonly leads to adverse effects at elevated drug levels.6 Since most drug therapy for ocular disease is administered topically, systemic metabolism is less of an issue, but genetic influence on drug metabolism in the eye should always be considered. However, when patients are non-responders, the more likely genetic mechanism is either a single or multiple polymorphism in the gene or genes responsible for the drug action. Genetics plays a central role in the etiology of many ocular diseases, and for some of these conditions there is mounting evidence that genetics also impacts ophthalmologists’ therapies and treatment strategies.
Pharmacogenetics in the Eye
Genetics of glaucoma is perhaps better understood than all but a handful of diseases.7 There is strong evidence linking specific forms of the disease with specific genotypes. For example, early-onset and normotensive glaucoma are linked to the myocilin gene, and congenital glaucoma is associated with cytochrome P1b1.
But what about linkage between therapies and specific genes? One paper reported an association between prostaglandin F2α receptor polymorphisms and intraocular pressure lowering in response to latanoprost,8 and another showed that SNPs of the b2 adrenergic receptors were associated with different responses to timolol.9 In both cases in the literature, differences were modest, yet they highlight the importance of a patient’s genetic contribution to therapeutic success and show how genetic testing in the future may help guide an ophthalmologist’s therapeutic decision-making process.
In the treatment of neovascular AMD, biologicals targeting vascular endothelial growth factor have become the first-line therapy in recent years. While the genetics of this particular disease aren’t as well characterized as in glaucoma, there is a strong association between AMD and SNPs of the complement cascade protein Complement Factor H.10 In addition, one CFH polymorphism is associated with a significantly lower response to anti-VEGF therapy.11 Other studies have suggested a genetic linkage between several gene products, including CFH, and in success rates with photodynamic therapy for AMD.
With this growing database of SNPs and associated treatment outcomes, it may be that age-related macular degeneration will be one of the first ocular disorders to employ genetic screening as part of a complete diagnostic workup.
Much less is known regarding the genetics of ocular allergy, although there are some studies focusing on the pharmacogenetics of asthma.12
There have been studies that explore pharmacogenomics of histamine metabolism, however, and these investigations have revealed clues regarding possible associations between the histamine-metabolizing enzymes and atopic disease.13 Histamine is normally metabolized via two pathways: conversion to N-methylhistamine by the enzyme histamine N-methyltransferase and conversion to imidazole acetaldehyde by the enzyme diamine oxidase (DAO; also known as histaminase).
One group of researchers identified SNP variants of DAO, and showed that patients with either atopic eczema or histamine intolerance are more likely to carry a DAO polymorphism associated with reduced enzyme activity; despite this, the researchers in the study concluded that this effect was not “sufficient to fully effectuate the disease state.”14,15 Such DAO polymorphisms could play a role in ocular allergy, since histaminase is known to be the primary pathway by which histamine is reduced following mast cell degranulation, and enzyme levels are known to be decreased in vernal keratoconjunctivitis.16 Interestingly, an SNP in another enzyme associated with histamine biosynthesis, histadine decarboxylase, has recently been reported to be associated with the risk of allergic rhinitis, although the underlying mechanism for such an effect remains unclear.17
The goal for the future is to use data from these SNP linkage studies to aid in determining which of the available therapies is likely to be the best for an individual in clinical practice. Conversely, pharmacogenetic and pharmacogenomic approaches can also aid in clinical trial design. Through pharmacogenomics, researchers may be able to eventually incorporate genetic screening in inclusion and exclusion criteria to improve the likelihood of clinically relevant responses. The downside of this screening, of course, is that it results in hastening the arrival of the day when doctors can no longer blame their patients when their prescribed medications don’t work.
Dr. Abelson, a clinical professor of ophthalmology at Harvard Medical School, consults in ophthalmic pharmaceuticals. Dr. McLaughlin is a medical writer at Ora Inc., in Andover.
1. Restasis [prescribing information]. Irvine, CA: Allergan Inc., 2009.
2. The International Dry-Eye Workshop. The Definition and Classification of Dry Eye Disease: Report of the Definition and Classification Subcommittee of the International Dry Eye Workshop. Ocul Surf 2007;5:2:75-92.
3. Snapinn SM, Jiang Q. Responder analysis and the assessment of a clinically relevant treatment effect. Trials 2007;8:31-37.
3. Diemunsch P, Leeser J, Feiss P, D’Hollander A, Bradburn BG, Paxton D, Whitmore J, Panouillot P, Navé S, Brown RA, Hahne WF. Intravenous dolasetron mesilate ameliorates postoperative nausea and vomiting. Can J Anaesth 1997;44:2:173-81.
4. Cornberg M, Hadem J, Herrmann E, Schuppert F, Schmidt HH, Reiser M, Marschal O, Steffen M, Manns MP, Wedemeyer H. Treatment with daily consensus interferon (CIFN) plus ribavirin in non-responder patients with chronic hepatitis C: A randomized open-label pilot study. J Hepatol 2006;44;2:291-301.
5. Relling MV, Giacomimi KM. Pharmacogenomics. In: L. Brunton, J Lazo, K Parker, I Buxton, D Blumenthal, eds. Goodman and Gilman’s Pharmacological Basis of Therapeutics, 11th edition. New York: McGraw Hill, 2006:Pages 93-98.
6. Lee NH. Pharmacogenetics of drug metabolizing enzymes and transporters: Effects on pharmacokinetics and pharmacodynamics of anticancer agents. Anticancer Agents Med Chem 2010;10:8:583-92.
7. John SW. Mechanistic insights into glaucoma provided by experimental genetics. Invest Ophthalmol Vis Sci 2005;46:2649-2661.
8. Sakurai M, Higashide T, Takahashi M, Sugiyama K. Association between genetic polymorphisms of the prostaglandin F2 receptor gene and response to latanoprost. Ophthalmology 2007;114:1039-45.
9. McCarty CA, Burmester JK, Mukesh BN, Patchett RB, Wilke RA. Intraocular pressure response to topical beta-blockers associated with an ADRB2 single-nucleotide polymorphism. Arch Ophthalmol 2008; 126:959-63.
10. Shastry BS. Genetic diversity and medicinal drug response in eye care. Graefes Arch Clin Exp Ophthalmol 2010;248:1057–1061.
11. Brantley MA Jr, Fang AM, King JM, Tewari A, Kymes SM, Shiels A. The association of complement factor H and LOC 387715 genotype with response of exudative age-related macular degeneration to intravitreal bevacizumab. Ophthalmology 2007;114:2168–2173.
12. Meyers DA. Genetics of asthma and allergy: What have we learned? J Allergy Clin Immunol 2010;126:3:439-46.
13. Garcia-Martin E, Ayuso P, Martinez C, Blanca M, Agúndez JAG. Histamine Pharmacogenomics. Pharmacogenomics 2010;10:5:867-883.
14. Maintz L, Benfadal S, Allam JP, Hagemann T, Fimmers R, Novak N. Evidence for a reduced histamine degradation capacity in a subgroup of patients with atopic eczema. J Allergy Clin Immunol 2006;117:1106–1112.
15. Maintz L, Yu C-F, Rodrıguez E, Baurecht H, Bieber T, Illig T, Weidinger S, Novak N. Association of single nucleotide polymorphisms in the diamine oxidase gene with diamine oxidase serum activities. Allergy 2011;66:893–902.
16. Abelson MB, Baird RS, Allansmith MR. Tear histamine levels in vernal conjunctivitis and other ocular inflammations. Ophthalmology 1980;87:8:812-4.
17. Gervasini G, Agundez JAG, Garcıa-Menaya J, Martınez C, Cordobes C, Ayuso P, Cornejo JA, Blanca M, Garcıa-Martın E. Variability of the L-Histidine decarboxylase gene in allergic rhinitis. Allergy 2010;65:1576–1584.