In ophthalmology, all of these elements contribute to pseudoexfoliative syndrome, a condition whose name is at the same time descriptive, historical and yet still a bit misleading. PXS is a systemic condition that first appears in the anterior chamber and can lead to cataract, glaucoma and complications with intraocular surgery. Here, we take a wide-ranging look at what the latest research reveals about PXS diagnosis, etiology, and genetics. We also compare PXS with related disorders and discuss ideas for treatment strategies.
What’s in a Name
The exfoliation syndrome associated with glaucoma that was first described in 1917 was later referred to as pseudoexfoliation to distinguish it from an occupational condition of “true exfoliation,” a delamination of the lens capsule common in glassblowers.1 In the medical literature of the early 20th century it was sometimes called senile exfoliation, and was considered a relatively rare form of secondary glaucoma restricted to those of advanced years and Nordic heritage. Even as recently as 2000, PXS was described as “a disease primarily of people of Scandinavian descent, with some features that suggest a genetic component.”2
|
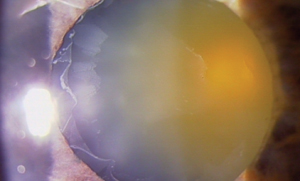
Diagnosis of PXS
The central finding in a diagnosis of PXS is the presence of white or light flakes in the anterior chamber of the eye, whose resemblance to epithelial debris gave rise to the description as exfoliation. Studies on the nature of the material suggest that it consists of connective tissues (elastin, collagen) together with adhered enzymes.4,5 Histologically, exfoliation material stains with periodic acid-Schiff reagents, indicating the presence of carbohydrates or proteoglycans. These properties led to the suggestion that the debris included amyloid proteins, and that it could be related to the central nervous system amyloid deposition that occurs in Alzheimer’s disease. It turns out there is no association between the two disorders (although PXS is linked to several systemic conditions; see below).6,7 While PXS is anecdotally associated with the Scandinavian population, the syndrome accounts for a greater percentage of open-angle glaucomas than primary disease in other ethnic populations, particularly Arabic, Mediterranean and Japanese patients.4,8,9 Worldwide, it is the most common cause of secondary glaucoma, and represents 5 to 18 percent of all open-angle disease. It’s estimated that, globally, 30 percent of people over age 60 have some form of anterior exfoliative deposits.2
Exfoliation syndrome goes beyond the build up of anterior segment debris. Changes in the lens capsule and iris are also seen in PXS.1-3 Reduced dilation function due to build up of PXS materials and degeneration of both sphincter and dilator muscles is often observed. In addition, focal membrane disruption in melanin-containing epithelial cells yields a pattern of peripupillary atrophy described as “moth eaten.”2,4
Collectively, PXS effects in the anterior segment lead to ocular hypertension and open-angle glaucoma. PXS is also associated with increases in angle closure, cataract and lens subluxation. Lens capsule atrophy in combination with poor mydriasis can make for challenging cataract surgery in PXS patients.2,4 While the combination of IOP-lowering medications and surgical approaches used to treat PXS is the same as that used for POAG, it’s possible that a better understanding of this glaucoma variant may lead to more fundamental progress in our knowledge of all forms of the disease.
PXS: A Systemic Disease
The association of PXS with connective tissue dysregulation is naturally more readily identifiable in the setting of the anterior segment. However, systemic manifestations of PXS have been investigated in both small-scale, retrospective studies and large-cohort, population-based research.10 As a disorder of connective tissues, particular interest has focused on the potential impact of the disease on cardiovascular function. Despite this interest, the first prospective, case-controlled studies have only been published in the past two to three years.
|

Several other conditions have been associated with PXS. An association with sensorineural hearing loss was shown in a study that compared auditory function in PXS patients with age-matched controls.13 The study examined both frequency range and threshold sensitivities. In contrast, the suggested association between PXS and Alzheimer’s disease has been discounted by a number of studies. Interest in this potential linkage stemmed from a desire to identify reliable predictive markers for the degenerative neurological disorder, but several studies have established that no linkage between the two disorders exists.6,7 Interestingly, a recent report suggested that deposition of β-amyloid in the lens may be the first reliable test for AD.14
The association of PXS with connective tissue dysregulation in the eye suggests that it may exert similar effects in other tissues, but for obvious reasons these effects are more readily identifiable in the setting of the anterior segment. Case studies have suggested associations between PXS and skin disorders, pulmonary disease and additional renal conditions. Progress in identifying the genetic loci for PXS is likely to advance investigations into these and other systemic diseases.15
Genetics of PXS and Glaucoma
Most of the genes linked to PXS have been identified with genome-wide association studies.16,17 One of the first such genes encodes one member of a ubiquitous family of enzymes involved in connective tissue metabolism, the lysyl oxidases. The LOXL1 gene product induces crosslinks between different sites on extracellular matrix proteins, and it’s thought that polymorphisms in the gene associated with PXS encode an altered enzyme resulting in too many crosslinks or inappropriate crosslinks, and ultimately a brittle, more easily damaged matrix.
GWAS have identified the LOXL1 locus as a PXS-associated gene in subjects from diverse genetic backgrounds, further strengthening the case for the gene as a key factor in PXS disease. Despite this, other loci including genes for the extracellular matrix protein, clusterin, the enzyme glutathione transferase and the signaling peptide TNF-α have also been implicated in PXS disease.8 Other factors, including unilateral presentation and variability in onset suggest that PXS etiology results from the combined effects of genetic predisposition coupled with one or more environmental factors.
Genetics of POAG are complex, and identification of glaucoma-associated genes such as myocilin or optineurin has provided some insight without yielding breakthroughs in either the etiology or treatment of the disease.18 While identification of genes involved in PXS may provide a clearer pathophysiology for the exfoliative aspects of the syndrome, we are still left with the daunting question of why some develop glaucoma while others do not. While lowering IOP remains the single best predictor of treatment success, patients with normal-tension glaucoma show that there’s more to it than that.
Research has found that the common denominator in all glaucoma-linked genes is an association with extracellular matrix and trabecular function, but these same genes may also be participants in homeostasis of the lamina cribrosa matrix. A unifying link, then, is a common effect on the extracellular matrix at both the front and the back of the eye.19,20
Changes in the signaling pathway regulated by TGFβ is another aspect of glaucoma pathophysiology seen in pseudoexfoliation and in all other forms of the disease. Patients with glaucoma of any etiology exhibit elevated aqueous humor TGFβ, a cytokine that both regulates matrix formation and causes increases in IOP.20, 21 Consistent with this role of TGFβ are two genetic disorders: Marfan syndrome and congenital scleroderma. Both of these conditions are due to a mutation of the microfibril-associated gene FBN1, both exhibit elevated TGFβ and both include glaucoma as part of the spectrum of their phenotypes. A recurring question in all of these observations regards cause and effect: Are elevated cytokines and elevated intraocular pressure responses to altered matrix dynamics, or are they somehow the initiators of these responses?
Putting PXS Pieces Together
Another intriguing piece of this puzzle is the phenomenon of normal-tension glaucoma, patients who exhibit the optic nerve head degeneration of glaucoma without an elevation in IOP. A growing body of evidence implicates a mismatch in trans-lamina cribrosa pressures in NTG.22,23 For some patients, normal ocular pressure could still yield differential pressures across the LC similar to those in patients with elevated IOP, if these patients had unusually low intracranial pressures. One condition in which this pressure difference at the LC may be an issue is idiopathic intracranial hypertension, a diagnosis associated with morbid obesity that is increasing worldwide.24 For patients with IIH, severe headache and optic disc swelling are the primary diagnostic features. These individuals have cerebrospinal fluid pressures that exceed normal IOP values. In severe cases, patients may require CSF shunting to reduce intracranial fluid pressures, but these devices typically yield extremely low intracranial fluid pressures, effectively “flipping” the trans-LC pressure differential. Patients with long-term in-dwelling shunts are at increased risk for glaucoma, and there is a recent prospective study showing that NTG patients have significantly lower CSF pressures than either controls or high-IOP glaucoma patients.25
One explanation for these observations is that factors such as elevated TGFβ are responses to the pressure differentials sensed at the level of the LC, the trabecular meshwork or both. Elevation of matrix regulatory stimuli may initiate a positive feedback cycle in which remodeling could promote or facilitate cupping, nerve damage and further remodeling. While speculative, this idea is consistent with current therapeutic standards that primarily slow the process down. An interesting idea might be to test a dual therapy of IOP-lowering agents in combination with antagonists of matrix remodeling factors such as TGFβ or connective tissue growth factor.26 This combination could, in theory, halt the remodeling cycle, reducing the effects of the TGFβ signaling pathway at both the front and the back of the eye.
This allows us to return to one of those inappropriately named diseases that we touched on earlier in this discussion, Marfan syndrome. Marfan is an autosomal dominant disorder caused by dysregulation of the gene for fibrillin 1, a connective tissue protein. Although it is still in the investigational stage, early clinical data suggest that a family of drugs called sartans (such as losartan) that are classified as angiotensin antagonists may be effective in reducing risk of aortic aneurysm, the major risk in patients with this syndrome.27 In addition to their action on the Renin-Angiotensin system, these drugs also act as physiological antagonists of TGFβ. For Marfan patients, the drug presumably attenuates excessive matrix synthesis in the walls of the great arteries. Patients with PXS, as well as those with POAG, might receive benefit from similar potential actions at the TM and LC. Perhaps calling sartans angiotensin inhibitors is yet another example of where an inaccurate name actually leaves out the most important, and most intriguing, characteristics of the drug. REVIEW
Dr. Abelson is a clinical professor of ophthalmology at Harvard Medical School. Dr. McLaughlin is a medical writer at Ora Inc. in Andover.
1. Johnson DH. The exfoliation syndrome: A continuing challenge. In: Albert DM and Jakobiec FA, ed. Principles and Practices of Ophthalmology, 2nd ed. Philadelphia: WB Saunders, 2000:2718-2730.
2. Ritch R, Schlötzer-Schrehardt U. Exfoliation syndrome. Surv Ophthalmol 2001;45:265-315.
3. Davis RE, Schuman JS. Pseudoexfoliation Syndrome: Don’t brush it off. Br J Ophthalmol.2013;97:1091-1092.
4. Schlötzer-Schrehardt U, Naumann GO. Ocular and systemic pseudoexfoliation syndrome. Am J Ophthal 2006;141:921-937.
5. Sein J, Galor A, Sheth A, et al. Exfoliation syndrome: New genetic and pathophysiologic insights. Curr Opin Ophthalmol 2013;24:2:167-74.
6. Ekström C, Kilander L. Pseudoexfoliation and Alzheimer’s disease: A population-based 30-year follow-up study. Acta Ophthalmol 2014;92:355-8.
7. Abramsson A, Landgren S, Zetterberg M, et al. No association of LOXL1 gene polymorphisms with Alzheimer’s disease. Neuromolecular Med 2011;13:2:160-6.
8. Elhawy E, Kamthan G, Dong CQ, Danias J. Pseudoexfoliation syndrome, a systemic disorder with ocular manifestations. Human Genomics 2012;6:22.
9. Olawoye OO, Pasquale LR, Ritch R. Exfoliation syndrome in sub-Saharan Africa. Int Ophthalmol 2014 May 21. [Epub ahead of print]
10. Cedrone C, Mancino R, Ricci F, et al. The 12-year incidence of glaucoma and glaucoma-related visual field loss in Italy: The Ponza eye study. J Glaucoma 2012;21:1:1-6.
11. Gonen KA, T Gonen T, Gumus B. Renal artery stenosis and abdominal aorta aneurysm in patients with pseudoexfoliation syndrome. Eye 2013;27:735–741.
12. Djordjevic-Jocic J, Jovanovic P, Bozic M, et al. Prevalence and early detection of abdominal aortic aneurysm in pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Curr Eye Res 2012;37:617-23.
13. Singham NV, Zahari M, Peyman M, et al. Association between ocular pseudoexfoliation and sensorineural hearing loss. J Ophthalmol 2014;2014:825936. Epub 2014 Apr 17.
14. Kerbage C, Sadowsky CH, Tariot PN, et al. Detection of amyloid β signature in the lens and its correlation in the brain to aid in the diagnosis of Alzheimer’s disease. Am J Alzheimers Dis Other Demen.2014 Feb 14. [Epub ahead of print]
15. Schlötzer-Schrehardt U. Molecular pathology of pseudoexfoliation syndrome/glaucoma – new insights from LOXL1 gene associations. Exp Eye Res;88:776-785.
16. Lemmela S, Forsman E, Sistonen P, Eriksson A, Forsius H, Jarvela I. Genome-wide scan of exfoliation syndrome. Invest Ophthalmol Vis Sci. 2007; 48:4136–4142.
17. Thorleifsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 2007;317:1397-1400.
18. Fingert JH. Primary open angle glaucoma genes. Eye (Lond) 2011;25:587-95.
19. Schlötzer-Schrehardt U, Hammer CM, Krysta AW, et al. LOXL1 deficiency in the lamina cribrosa as candidate susceptibility factor for a pseudoexfoliation-specific risk of glaucoma. Ophthalmology 2012;119:9:1832-43.
20. Kuchtey J, Kuchtey RW. The microfibril hypothesis of glaucoma: Implications for treatment of elevated intraocular pressure. J Ocul Pharm Ther 2014;30:170-180.
21. Fuchshofer R. The pathogenic role of transforming growth factor-β2 in glaucomatous damage to the optic nerve head. Exp Eye Res 2011;93:2:165-9.
22. Fleischman D, Allingham RR. The role of cerebrospinal fluid pressure in glaucoma and other ophthalmic diseases. Saudi J of Ophthalmol 2013;27:97–106.
23. Jonas JB, Wang N, Wang YX, et al. Ocular hypertension: General characteristics and estimated cerebrospinal fluid pressure. The Beijing eye study 2011. PLoS One 2014;9:7:e100533.
24. Wakerly BR, Tan MH, Ting EY. Idiopathic Intracranial hypertension. Cephalalgia. 20 May 2014. pii: 0333102414534329. [Epub ahead of print].
25. Ren R, Jonas JB, Tian G, et al. Cerebrospinal fluid pressure in glaucoma: A prospective study. Ophthalmology 2010;117:2:259-66.
26. Wallace DM1, Clark AF, Lipson KE, Andrews D, Crean JK, O’Brien CJ. Anti-connective tissue growth factor antibody treatment reduces extracellular matrix production in trabecular meshwork and lamina cribrosa cells. Invest Ophthalmol Vis Sci 2013;54:13:7836-48.
27. Matt P, Eckstein F. Novel pharmacological strategies to prevent aortic complications in Marfan syndrome. J Geriatr Cardiol 2011;8:4:254-7.


