In 1971, Morton F. Goldberg, MD, published the now commonly used sickle cell retinopathy grading system based upon the distinct pattern of retinal vascular remodeling in the peripheral retina in patients with sickle cell disease. Over the years, this grading system has helped us understand the natural history of retinal vascular changes in sickle cell patients. Though modern advances in pharmacotherapies and surgical techniques have helped us improve visual outcomes, there’s still a lack of Level 1 evidence to inform our management of sickle cell retinopathy.
In this article, we’ll highlight knowledge gaps in SCR, discuss preferred practice patterns and analyze several illustrative cases of SCR management dilemmas.
The Grades of Sickle Cell
Dr. Goldberg’s observations were derived from careful clinical exams using indirect ophthalmoscopy, 30-degree standard field fundus photography and fluorescein angiography with peripheral sweeps in patients with SCR.1 Following is a brief description of the grading system.
In Goldberg Stage 1, peripheral arteriolar occlusions are observed, which can then progress to Stage 2, arterio-venous anastomoses. Goldberg Stage 3 marks the onset of proliferative sickle retinopathy, in which arterio-venous connections proliferate into characteristic fan shaped complexes called “sea-fan” neovascularization, so named for their resemblance to the marine animal, Gorgonia flabellum. Patients with sickle cell disease generally maintain good vision until they progress to Goldberg Stage IV disease, when vitreous hemorrhage occurs due to vitreous contraction on sea-fan neovascular complexes. The avascular peripheral retina in SCD is thin and therefore prone to retinal breaks. Vitreous traction may cause sea-fan neovascularization to exert traction on the peripheral retina, resulting in Stage V (tractional or combination tractional/rhegmatogenous retinal detachment), and necessitating surgical repair.
Though this staging system for SCR is informed by peripheral retinal changes, Dr. Goldberg and his colleagues also described macular vascular changes including microaneurysm-like dots, dark and enlarged segments of terminal macular arterioles, and hairpin-shaped venular loops with slit lamp and indirect biomicroscopy.2 Contemporary advances in retinal imaging including optical coherence tomography, optical coherence tomography angiography, ultra-widefield fundus photography and ultra-widefield fluorescein angiography have provided a greater understanding of macular and peripheral anatomy of the retina that contributes to pathophysiology of SCR.
How Retinopathy Relates to SCD
The most common causes of vision loss in SCD are sequelae of PSR, e.g., vitreous hemorrhage and/or retinal detachment. In our patients with diabetes, we’re able to link poor glycemic control with the risk of developing sight-threatening proliferative retinopathy, and counsel the patient and referring primary care provider accordingly. In SCD, we still don’t understand the correlation between the patient’s systemic hemoglobinopathy and their risk of sight-threatening eye disease.
 |
Genotype is the risk factor most strongly associated with development of PSR. PSR occurs earlier and more commonly in HbSC disease; it’s noted in about 43 percent of HbSC subjects as compared with 14 percent of HbSS subjects in a Jamaican cohort study.3 Though mechanisms for this disparity in PSR between HbSS and HbSC disease aren’t completely understood, blood viscosity and overall circulation and transit time of abnormal hemoglobin cells may play a role. A hemoglobin S molecule results from a valine substitution for a glutamine, and the red cell survival time in HbS is about seven to 14 days. HbS polymerizes in the deoxygenated state. A hemoglobin C molecule results from a lysine substitution for a glutamine. HbC molecules crystalize within red cells leading to higher blood viscosity in patients with HbSC disease. HbC red cells survive about 40 days. In HbSC, patients have a higher hematocrit and, therefore, also have a higher volume of abnormal hemoglobin molecules, decreased blood flow and longer vascular transit time due to increased sludging of blood in the microvasculature.
This sluggish blood flow results in prolonged hypoxia, and indolent release of pro-angiogenic growth factors such as vascular endothelial growth factor, promoting pathologic neovascularization. In HbSS, there may be more overall complete vaso-occlusions with obliterations of microcapillaries in the retinal circulation and, therefore, more complete areas of anoxia. Interestingly, SCR is quite heterogeneous in its presentations, and its phenotypic appearance can differ significantly, even across SCD patients with the same genotype.
Evidence of SCD vaso-occlusions within the retinal circulation in patients with SCD have been observed in patients as early as 6 months old, after the protective effects of fetal hemoglobin (HbF) abate. These vaso-occlusions in the microvasculature are cumulative over a lifetime. Age is another risk factor for PSR, with the highest rates of PSR progression between ages 20 to 39. We typically don’t see active PSR or PSR progression in SCD patients over age 50, and in patients this age, much of the SCR we see has a “burnt out” fibrotic appearance of PSR neovascular complexes. Male sex and PSR in the contralateral eye are also risk factors for PSR development.3 Persistent fetal hemoglobin, hydroxyurea use (increases blood flow and raises HbF), and chronic transfusions appear to be protective factors for SCR development.
Patients with SCD may have myriad medical complications from their hemoglobinopathy, including recurrent vaso-occlusive pain crises, strokes, silent cerebral infarcts, pulmonary hypertension, avascular necrosis of the bones and nephropathy. Further study is required to better understand how the hemoglobinopathy affects a patient with SCD and his or her risk for vision loss from retinal nonperfusion or retinal vascular occlusion or infarction, incidence of PSR or risk of PSR progression, and how these correlate with any of these other sequelae of SCD-related end-organ damage.
Screening for SCD Patients
Current sickle cell retinopathy screening guidelines are based only on expert consensus and strong recommendations, but low-quality evidence.4 Recommendations include “referral to an ophthalmologist for a dilated eye examination to evaluate for retinopathy beginning at age 10,” and “rescreen at one- to two-year intervals for persons having a normal dilated retinal examination.”4 Even though these recommendations aren’t based on strong evidence, they’re reasonable in that, as previously mentioned, it’s unusual to observe PSR in children under the age of 10 with SCD, and the highest rates of PSR progression typically occur in the time frame from adolescence to 30 years of age.
One four-year, retrospective, observational cohort study further validated the expert consensus recommendations by evaluating optimal timing for sickle cell retinopathy screening.5 The study’s authors concluded screening for SCR in asymptomatic children could reasonably take place at age 9 for children with HbSC and at age 13 for children with HbSS. From my perspective, this recommendation is very similar to the NIH expert consensus paper, and screening at age 10 for all patients with all SCD genotypes appropriately captures the intent of this guideline.
In our clinics, we encourage referral of children with SCD as early as age 5, because although they’re unlikely to have developed PSR, at this age even subtle microvascular occlusive disease can be identified with detailed retinal exams and imaging, including OCT and OCTA. Young children can be reasonably cooperative with the retinal exam and imaging at this age.6 In partnership with our hematology colleagues, we find it helpful to discuss screening expectations for annual retinal examinations with pediatric SCD patients and their families, so, ideally, these surveillance visit patterns can be ingrained in these patients starting at a young age and can then continue for the rest of our patients’ lives.
Managing PSR
PSR is typically treated to avoid disease progression in to Goldberg Stage 4 or Stage 5 disease. Scatter laser is the mainstay of treatment, while anti-VEGF therapy can be a useful adjunctive therapy.
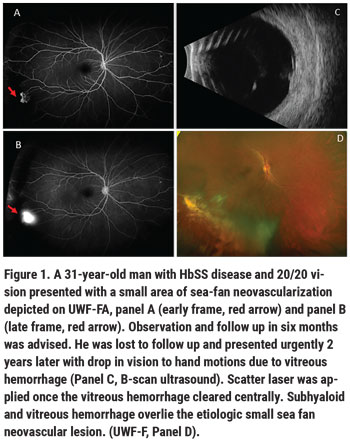
• Scatter laser photocoagulation. When screening for SCR, the objective is to identify pathologic neovascularization so treatment can be considered, or if treatment is deferred, these lesions can documented and followed closely over time. PSR is unique among the proliferative retinopathies, as sea-fan neovascular complexes tend to auto-infarct and fibrose without visual consequence in upwards of 30 percent of eyes without visual consequence.3 Laser photocoagulation remains the first line treatment. We recommend scatter laser treatment for patients with large, vascularized sea fan lesions; enlarging sea fans; monocular patients and/or those with advanced PSR in the contralateral eye; and in patients in whom follow-up may not be reliable (Figure 1).
 |
Laser photocoagulation has been shown to slightly decrease incidence of vitreous hemorrhage compared to observation in control eyes with PSR.7 Laser photocoagulation hasn’t been shown to decrease incidence of new sea-fan neovascularization and appears to have minimal effect on induction of sea-fan auto-infarction.7 Therefore, small sea-fan neovascular complexes can be observed. We recommend using UWF-FA to guide scatter laser treatment. We apply laser to barricade sea-fan neovascular complexes, to the areas of surrounding peripheral retinal ischemia and to the transitional zone between perfused and non-perfused retina. Using previously described techniques based on the location of angiogenic growth factors identified in a study of autopsy eyes with PSR, we extend treatment just posterior to this transitional zone border.8 Peripheral retinal ischemia has been shown to be progressive in SCR, however, we don’t perform scatter laser for retinal ischemia in the absence of neovascularization.
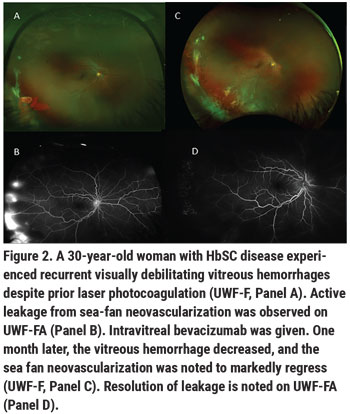
• Anti-VEGF injection therapy. Anti-VEGF medications are a helpful adjunct to scatter laser in PSR. Small case series have shown intravitreal bevacizumab to be helpful in facilitating resolution of vitreous hemorrhage when the hemorrhage prevents adequate laser initiation or supplementation; these injections can also be effective in decreasing vascular leakage and recurrent vitreous hemorrhage in PSR (Figure 2).9 Intravitreal bevacizumab has also proven to be a useful preoperative medication, causing regression of active sea-fan neovascular complexes and facilitating their dissection from the retinal surface, which decreases the risk of intraoperative bleeding in vitrectomy for retinal detachment repair. We don’t yet have a defined optimal treatment paradigm for anti-VEGF injection for PSR management, however. We tend to treat as needed based upon vascularization of sea-fan complexes on clinical exam and UWF-FA, and upon the extent of sea-fan leakage on UWF-FA. Vitreous hemorrhages from PSR tend to significantly improve within 4 to 6 weeks with observation and with anti-VEGF treatment. Given complexities of vitreoretinal surgery and perioperative management in patients with SCD (discussed in the next section), we highly recommend observation and medical management of PSR vitreous hemorrhage in the absence of retinal detachment when possible.
• Surgery for PSR. It’s important to recognize patients with PSR neovascularization may have an undiagnosed mild hemoglobinopathy, and may present with vision loss due to vitreous hemorrhage or RD in the absence of known clinical history of SCD. Serum electrophoresis and referral for hematologic work up should be obtained when patients present with retinal findings consistent with PSR in the absence of known hemoglobinopathy.
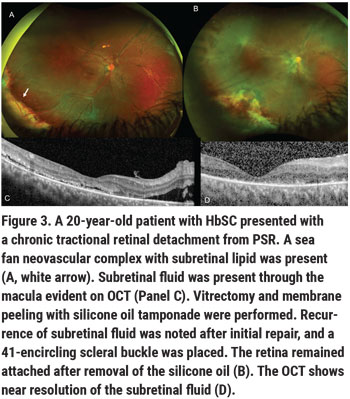 |
Surgical repair should be reserved for eyes with vision-threatening eye disease for which medical management options have been exhausted. Surgical indications in PSR can include non-clearing vitreous hemorrhage (particularly in a monocular patient), retinal detachment, symptomatic epiretinal membrane, macular hole or vitreomacular traction. The peripheral ischemic retina is thin, therefore eyes with SCD are prone to retinal breaks and rhegmatogenous retinal detachments, as well as tractional or combined rhegmatogenous/tractional retinal detachments. Epimacular proliferation may occur either by natural history or following scatter laser photocoagulation, resulting in symptomatic epiretinal membrane with macular pucker, macular hole or vitreomacular traction. Even when modern vitrectomy techniques such as widefield viewing to maximize retinal visualization, small-gauge instruments and valved cannulas to provide tight control of intraocular pressure are used, PSR eyes with RD are prone to iatrogenic breaks and may show complication rates of up to 50 percent.10 Preoperative communication with the patient’s hematologist is important to identify the possible role of preoperative exchange blood transfusion, and for the management of any anti-coagulation therapy. Communication with the anesthesiologist during the surgery is also critical, taking care to maximize the patient’s perioperative hydration, temperature, oxygenation and analgesia. We prefer general anesthesia for our SCD patients. Local anesthesia with sub-Tenon’s block can be considered if general anesthesia is contraindicated. Care must be taken to monitor IOP, avoiding the use of a retrobulbar block to decrease the risk of central retinal artery occlusion. We use a bimanual surgical technique for dissection of preretinal membranes with chandelier illumination, try to employ segmentation rather than delamination techniques, and minimize any tension on the retina to avoid causing iatrogenic breaks.
Historically, scleral buckles were avoided in patients with SCD given the risk of anterior segment ischemia when high, broad scleral buckles were used. In light of the fact that patients with PSR-retinal detachment are typically young and phakic—with incomplete posterior hyaloid separation and possible inferior vitreous base pathology—low, narrow encircling scleral buckles or segmental buckles may be used, and we’ve found them to be beneficial in PSR surgery (Figure 3).
 |
Other Causes of Vision Loss
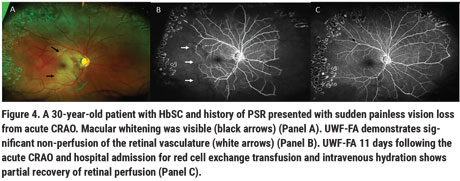
Advanced-stage PSR is the most common cause of significant vision loss in SCD, but sudden vision loss can also rarely occur from acute onset vascular occlusions in the posterior pole, central or branch retinal artery occlusion (RAO) or acute macular infarction.
Retinal artery occlusion is more common in the HbSS but can occur in HbSC (Figure 4). Many patients with SCD have chronic macular flow voids on OCTA, most noted in the deep retinal capillary vascular plexus to a greater extent than in the superficial plexus. These flow voids are commonly observed in the temporal macular region, a vascular watershed zone. Children with SCD were noted to have decreased macular vascular density but similar retinal thicknesses when compared to age- and race-matched controls unaffected by the disease.6 Structural OCT macular thinning has also been observed in adults with SCD, suggesting that vascular occlusions precede structural retinal thinning. The visual significance of these macular flow voids remains unclear, and these patients are thought to be visually asymptomatic. However, microperimetry studies in SCD observed central scotomas that correspond to these areas of structural thinning.13 Patients with these macular flow voids and corresponding areas of OCT thinning may have chronic subclinical near-vision deficits that we don’t pick up on in our vision testing, as we don’t routinely check near vision on these patients.
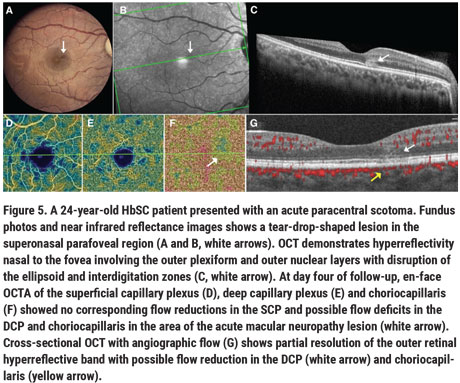
Rarely, acute vision loss with sudden onset of central scotomas has also been noted in patients with SCD from paracentral acute middle maculopathy (PAMM) or acute macular neuroretinopathy (AMN). PAMM likely occurs due to vascular flow impairment in the deep and/or intermediate retinal capillary plexus, and has been hypothesized to precede macular thinning in sickle cell maculopathy.14,15 The deep capillary plexus may be particularly susceptible to ischemia given its high oxygen demand, and because its vascular supply occurs in a watershed zone of the retinal circulation. Ischemic insult to the deep capillary plexus ischemia and possibly the choroid may result in AMN, often noted as a tear-drop-shaped, paracentral, hyperreflective lesion well-visualized on infrared OCT (Figure 5).16
It follows that given their predisposition to capillary occlusion and red-blood-cell sickling, patients with SCD would be vulnerable to these episodes of sudden vision loss. In our institution, when SCD patients experience sudden vision loss attributed to vascular occlusions, these events are treated as acute neurologic events, and these patients are referred for urgent hematologic consult, hydration, oxygenation and possible exchange transfusion. AMN and PAMM lesions typically resolve when followed over time, though the natural course may be improved with systemic optimization of SCD through hydration, oxygenation and exchange transfusion. However, though these patients typically complain of persistent central or paracentral scotomas, even the acute infarct resolves and perfusion improves. Interestingly, these events have been reported to occur in various SCD genotypes, and don’t necessarily correlate with an individual’s overall systemic SCD morbidity.
A Look Toward the Future
Though our knowledge of the pathophysiology, natural history and best practices in surgical and medical management of SCR has grown tremendously since the initial Goldberg classification was published in 1971, there remains much about SCR we still don’t know. Fortunately, over recent years, there’s been a heightened interest in sickle-cell disease and in sickle-retinopathy research, including an exponential increase in the number of publications evaluating retinal imaging in SCD—more in the past 10 years than in the previous four decades combined. Along those lines, in our retina practice we’re working to establish evidence-based practice patterns for SCR, using retinal imaging to incorporate not only peripheral retinal findings, but sickle maculopathy as well, with the ultimate goal being an update to the staging classification system.
 |
It’s an exciting time for our patients with SCD and for us clinicians involved in their care. For years, hydroxyurea was the only FDA-approved medication for the treatment of SCD. Now, there are newer pharmacotherapies for SCD treatment, such as crizanlizumab (Adakveo, Novartis), a p-selectin inhibitor intravenous infusion shown to decrease the number of painful vaso-occlusive crises; and voxelotor (Oxbryta,Global Blood Therapeutics), an oral medication shown to increase oxygen affinity to hemoglobin, inhibit red cell polymerization and reduce hemolysis.
Systemic therapies such as bone-marrow transplantation and CRISPR gene editing11 have also had success in altering the disease course in SCD, and the hope is that these therapies will ultimately be curative. It’ll be fascinating to observe the effects of these systemic therapies on SCD end-organ damage, including sickle-cell maculopathy and peripheral retinopathy. Unfortunately, many of these treatments aren’t yet available to most patients with SCD due their cost and their potential systemic toxicity (chemotherapy is required prior to bone-marrow transplantation and gene therapy).
Further, SCD affects approximately 100,000 Black Americans, many of whom are from medically underserved communities and facing daily struggles related to their disease care. For these patients, limited or no access to and/or mistrust of the medical community due to past ethical violations may prevent them from getting these treatments.12 Because of these issues, in addition to understanding and treating hemoglobinopathy and end-organ damage in patients with SCD, we must also address racial disparities in health care so that this population can truly benefit from these groundbreaking therapies. REVIEW
Dr. Scott is an associate professor of ophthalmology in the retina division of the Wilmer Eye Institute at Johns Hopkins University School of Medicine.
1. Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol 1971;71;3:649-65.
2. Stevens TS, Busse B, Lee CB, et al. Sickling hemoglobinopathies; macular and perimacular vascular abnormalities. Arch Ophthalmol 1974;92:455-463.
3. Downes SM, Hambleton IR, Chuang EL, et al. Incidence and natural history of proliferative sickle cell retinopathy: Observations from a cohort study. Ophthalmology 2005;112:1869-1875.
4. Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033-1048.
5. Li J, Bender L, Shaffer J, et al. Prevalence and onset of pediatric sickle cell retinopathy. Ophthalmology 2019;126:1000-1006.
6. Ong SS, Linz MO, Li X, et al. Retinal thickness and microvascular changes in children with sickle cell disease evaluated by optical coherence tomography (OCT) and OCT angiography. Am J Ophthalmol 2020;209:88-98.
7. Farber MD, Jampol LM, Fox P, et al. A randomized clinical trial of scatter photocoagulation of proliferative sickle cell retinopathy. Arch Ophthalmol 1991:109:363-367.
8. Rodrigues M, Kashiwabuchi F, Deshpande M, et al. Expression pattern of HIF-1α and VEGF supports circumferential application of scatter laser for proliferative sickle retinopathy. Inves Ophthalmol Vis Sci 2016;57:6739-6746.
9. Cai CX, Linz MO, Scott AW. Intravitreal bevacizumab for proliferative sickle retinopathy: a case series. J of VitRet Dis 2018;2:32-38.
10. Chen RWS, Flynn HW, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: A case series. Am J Ophthalmol 2014;157:870-875.e1.
11. Marcus AD. Gene editing shows promise in sickle cell disease. The Wall Street Journal. Dec 7, 2020.
12. Farook F, Mogayzel PJ, Lanzkon S, et. Al. Comparison of US federal and foundation funding of research for sickle cell disease and cystic fibrosis and factors associated with research productivity. JAMA Netw Open 2020;3:3:e201737.
13. Chow CC, Geneat MA, Anastasakis A, et al. Structural and functional correlation in sickle cell retinopathy using spectral-domain optical coherence tomography and scanning laser ophthalmoscope microperimetry. Am J Ophthalmol 2011;152:704-711.e2.
14. Jung JJ, Chen MH, Frambach CR, Rofagha S, Lee SS. Spectral domain versus swept source optical coherence tomography angiography of the retinal capillary plexuses in sickle cell maculopathy. Retin Cases Brief Rep. 2018;12:2:87-92.
15. Hussnain SA, Coady PA, Stoessel KM. Paracentral acute middle maculopathy: Precursor to macular thinning in sickle cell retinopathy. BMJ Case Rep 2017;2017.
16. Ong SS, Ahmed I, Scott AW. Association of acute macular neuroretinopathy or paracentral acute middle maculopathy in sickle cell disease. Ophthalmology Retina 2020 (in press).


