| ||||||
Pathophysiology
Mutations in NDP, a gene coding for norrin, have been implicated in diseases involving retinal vasculogenesis, including Coats’ disease.2 A “Coats’-like” retinopathy has been observed in genetic syndromes such as autosomal dominant facioscapulohumeral muscular dystrophy (Hallermann-Streiff syndrome) and familial renal-retinal dystrophy (Senior-Loken syndrome).3,4 Despite these associations, sporadic cases are the rule, and no hereditary pattern has been consistently identified.
Histopathologic studies of Coats’ specimens demonstrate pericyte loss, which allows for the formation of aneurysms. Breakdown of the endothelial blood-retinal barrier causes leakage into the vessel wall, leading to dilation and telangiectasis. This pathologic mechanism is similar to that observed in diabetic retinopathy, which has implications in treatment.5,6
|
Clinical Presentation
The majority of Coats’ disease is diagnosed between ages 8 and 16.1 There are reports of cases presenting in adulthood, but this is less common.7,8 Greater than 75 percent of patients are male, and 95 percent of cases are unilateral.1 Alternative diagnoses should especially be considered in female, adult-onset or bilateral cases.
|
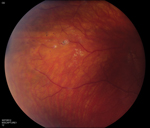
In 2000, a classification system was proposed using funduscopic features of the disease (See Table 2).11 Retinal vessel telangiectasia and aneurysmal dilations (See Figure 1) are the hallmark of stage 1, and are most commonly noted anterior to the equator in the temporal and inferior quadrants. Fusiform arteriolar dilatation (“light bulb” aneurysms) and venous sheathing or beading may occur (See Figure 2). The presence of exudates indicates stage 2, and stage 3 includes both subtotal and total retinal detachment. Pre- and subretinal fibrosis may be present in advanced disease. Cases with secondary glaucoma and pre-phthisis bulbi are classified as stage 4 and 5, respectively. The natural history is highly variable but progression is generally expected. Staging can be helpful in monitoring disease activity and response to treatment. Late reactivation is not uncommon and lifetime surveillance is prudent.
| ||||||||||||||||||||||||||||||||||||||||||||||||
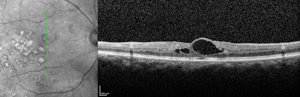
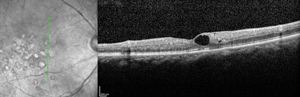
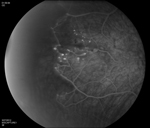
Ophthalmoscopy is often sufficient to make the diagnosis of Coats’ disease. Examination under anesthesia may be necessary in young children. Fundus photography is useful at baseline and on subsequent follow-up visits to monitor for progression and identify new areas of disease activity. Fluorescein angiography provides optimal visualization of vascular abnormalities which hyperfluoresce early and leak late (See Figure 1), helping target treatment. Optical coherence tomography, including intraoperative techniques,12 has been reported as useful in localizing subtle intraretinal edema or subretinal fluid, as well as monitoring response to therapy.13,14 In advanced disease with substantial subretinal fluid, B scan ultrasound may be helpful to rule out an underlying mass lesion.15 CT and MRI may also have some utility in atypical cases to identify calcification or enhancement that may be indicative of retinoblastoma.
Treatment
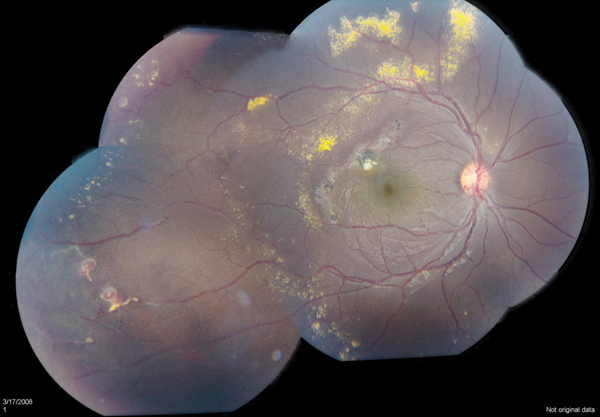
Many patients develop significant retinal detachment with exudates, which have a predilection for the macula, possibly due to currents of subretinal fluid created by activity of the pump mechanism of the retinal pigment epithelium. Such deposits have high concentrations of protein, cholesterol, hemosiderin-laden macrophages, and RPE cells with fibrous metaplasia.16,17 This can lead to subfoveal fibrotic nodules with a particularly poor prognosis. Although observation may be appropriate in some very limited peripheral stage 1 Coats’ disease, early ablative or combination therapy can limit exudation and, ultimately, prevent vision loss.
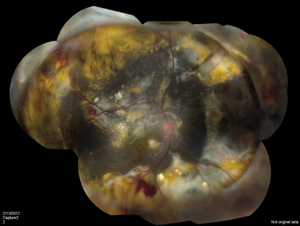
Laser photocoagulation is the modality of choice for mild to moderate disease with exudation. In the Shields’ classification system proposed in 2000, stage 1 and 2, leaking telangiectases and aneurysms, can be directly treated with good results (See Figure 3).18,19 Scatter or barricade strategies are generally less efficacious. Multiple sessions are often necessary as new lesions become clinically evident.1
If there is significant subretinal fluid, as in stage 3 disease, cryotherapy may be required. Exudation can increase immediately after treatment, especially if a substantial area is involved.11 Retinal contracture and folds may also occur, and it is generally recommended that only two quadrants be treated at a time, and that sessions be separated by at least one month. Laser photocoagulation to barricade fluid prior to cryotherapy may help limit progression of subretinal fluid.20
The era of intravitreal injections has yielded several new options for managing the exudative complications of Coats’ disease. In the absence of prospective comparative studies, it is best to consider pharmacotherapy as an adjuvant to ablative techniques. Combination therapy (e.g., intravitreal triamcinolone or anti-vascular endothelial growth factor agents before or at the time of ablation) offers the potential benefit of immediate stabilization (or improvement) with long-term control.
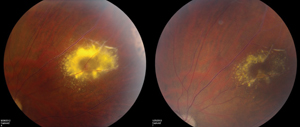
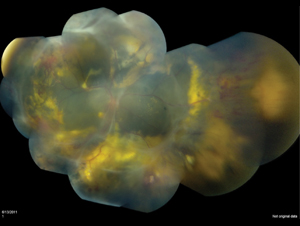
Yu-Guang He, MD, and colleagues reported a mean vitreous VEGF level of 2,394.5 pg/ml in Coats’ patients compared with 15.3 pg/ml in controls.21 Although the exact pathophysiologic mechanism needs further study, there seem to be therapeutic implications. In the past six years, 19 authors have published cases or series of anti-VEGF therapy for Coats’ disease in stages 2 to 4. Nearly all have shown positive results with respect to visual or anatomic outcomes (See Figure 4). In a recent retrospective review with age-matched controls, intravitreal bevacizumab plus thermal ablation was shown to have fewer treatment failures over laser alone.22 Although most reports are overwhelmingly favorable, some have cautioned of increased vitreoretinal traction following intravitreal anti-VEGF therapy in stages 2 to 3 Coats’ disease.23
Intravitreal triamcinolone may be especially beneficial in eyes with total bullous exudative detachment (TBERD, stage 3b).24 Total resolution of subretinal fluid has been reported with a single injection.25 The risks and benefits should be carefully considered since cataract, elevations in intraocular pressure and other injection-related complications are all possibilities. We generally prefer administration of 2 mg preparations of IVT over 4 mg, as the former is associated with a lower rate of elevated IOP.
Surgical intervention is rarely necessary and is reserved for late stage or refractory disease. In select cases, patients with extensive retinal detachment may benefit from vitrectomy. In particular, patients with pre-retinal membranes or contribution from tractional components may require membrane peel (See Figure 5). Enucleation may be necessary in stage 4 and 5 disease, where a blind, painful eye may develop.
More than a century has passed since the original description of Coats’ disease. Our understanding of the underlying pathophysiology continues to improve, as do our diagnostic and treatment modalities. Intravitreal therapy represents an exciting addition to traditional ablative techniques. Early identification of exudation and appropriately aggressive intervention can modify the course of the disease and improve patient outcomes.
Dr. Pitcher is a second year fellow at Wills Eye Institute and a clinical instructor of ophthalmology at Thomas Jefferson University School of Medicine. Dr. Regillo is director of the Retina Service at Wills and a professor of ophthalmology at Jefferson.
1. Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats’ disease in 150 cases: The 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001;131:561-571.
2. Black GC, Perveen R, Bonshek R, Cahill M, Clayton-Smith J, et al. Coats’ disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: A role for norrin in retinal angiogenesis. Hum Mol Genet 1999;8(11):2031-2035.
3. Newell SW, Hall BD, Anderson CW, Lim ES. Hallermann-Streiff syndrome with Coats’ disease. J Pediatr Ophthalmol Strabismus 1994;31:123-125.
4. Schuman JS, Lieberman KV, Friedman AH, Berger M, Schoeneman MJ. Senior-Loken syndrome (familial renal-retinal dystrophy) and Coats’ disease. Am J Ophthalmol 1985;100:822-827.
5. Egbert PR, Chan CC, Winter FC. Flat preparations of the retinal vessels in Coats’ disease. J Pediatr Ophthalmol 1976;13:336-339.
6. Fernandes BF, Odashiro AN, Maloney S, Sajdenweber ME, et al. Clinical-histopathological correlation in a case of Coats’ disease. Diagn Pathol 2006;1:24.
7. Smithen LM, Brown GC, Brucker AJ, Yannuzzi LA, et al. Coats’ disease diagnosed in adulthood. Ophthalmology 2005;112:1072-1078.
8. Shienbaum G, Tasman WS: Coats’ disease. A lifetime disease. Retina 2006;26:422-424.
9. Budning AS, Heon E, Gallie BL. Visual prognosis of Coats’ disease. J AAPOS 1998;2(6):356-359.
10. Shields CL, Uysal Y, Benevides R, Eagle RC Jr, et al. Retinoblastoma in an eye with features of Coats’ disease. J Pediatr Ophthalmol Strabismus 2006;43:313-315.
11. Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: The 2000 Proctor Lecture. Am J Ophthalmol 2001;131:572-583.
12. Henry CR, Berrocal AM, Hess DJ, Murray TG. Intraoperative spectral-domain optical coherence tomography in Coats’ disease. Opthalmic Surg Lasers Imaging 2012 Jul 26;43:e80-84.
13. Kessner R, Barak A, Neudorfer M. Intraretinal exudates in Coats’ disease as demonstrated by spectral-domain OCT. Case Rep Ophthalmol 2012;3(1):11-15.
14. Jun JH, Kim YC, Kim KS. Resolution of severe macular edema in adult Coats’ disease with intravitreal triamcinolone and bevacizumab injection. Korean J Ophthalmol 2008;22(3):190-193.
15. Atta HR, Watson NJ. Echographic diagnosis in advanced Coats’ disease. Eye 1992;6:80-85.
16. Hsu J, Forbes B, Maguire AM. Total exudative retinal detachment in Coats’ disease. Biochemical analysis of the subretinal exudate. Retina 2006;26:831-833.
17. Khurana RN, Samuel MA, Murphree AL, Loo RH, et al. Subfoveal nodule in Coats’ disease. Clin Exp Ophthalmol 2006;33(3):301-302.
18. Couvillion SS, Margolis R, Mavrofjides E, Hess D, Murray TG. Laser treatment of Coats’ disease. J Pediatr Ophthalmol Strabismus 2005;42:367-368.
19. Scheffler AC, Berrocal AM, Murray TG. Advanced Coats’ disease. Management with repetitive aggressive laser ablation therapy. Retina 2008;28:38-41.
20. Budning AS, Heon E, Gallie BL. Visual prognosis of Coats’ disease. J AAPOS 1998;2(6):356-359.
21. He YG, Wang H, Zhao B, Lee J, Bahl D, McCluskey J. Elevated vascular endothelial growth factor level in Coats’ disease and possible therapeutic role of bevacizumab. Graefe’s Arch Clin Exp Ophthalmol 2010;248(10):1519-21.
22. Ray R, Baranano DE, Hubbard GB. Treatment of Coats’ disease with intravitreal bevacizumab. Br J Ophthalmol 2013;97(3)272-277.
23. Ramasubramanian A, Shields CL. Bevacizumab for Coats’ disease with exudative retinal detachment and risk of vitreoretinal traction. Br J Ophthalmol 2011;96(3):356-359.
24. Othman IS, Moussa M, Bouhaimed M. Management of lipid exudates in Coats disease by adjuvant intravitreal triamcinolone: Effects and complications. Br J Ophthalmol 2010;94:606-610.
25. Ghazi NG, Al Shamsi H, Larsson J, Abboud E. Intravitreal triamcinolone in Coats’ disease. Ophthalmology 2012;119(3):648-649.


