Neuro-ophthalmology was consulted, and a magnetic resonance imaging and magnetic resonance angiogram of the brain and orbits with and without intravenous gadolinium contrast was recommended. An inflammatory workup, including a complete blood count, erythrocyte sedimentation rate, rapid plasma reagin, fluorescent treponemal antibody absorption, anti-neutrophil cytoplasmic antibody panel and Lyme titer was initiated. Chest X-ray revealed a prominent left inferior hilum. She was promptly admitted to the Wills Eye Neuro-ophthalmology inpatient service.
|
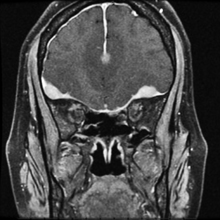
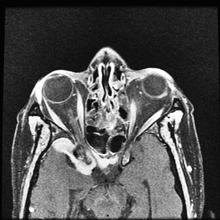
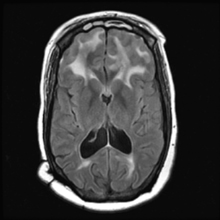
The MRI/MRA revealed pachymeningial enhancement (See Figure 3) and lesions compressing the optic nerves bilaterally and cranial nerve III on the right side (See Figure 4). The lesions were isointense on T1 and hypointense on T2. There was also diffuse white matter hyperintensity on T1 FLAIR images in the region of the frontal lobes (See Figure 5), which could possibly account for her relative indifference about her significant vision loss. No further workup was pursued given significant improvement on systemic steroids, her known history of biopsy-proven sarcoidosis, and imaging consistent with neurosarcoidosis.
Two months after discharge, the patient remained NLP in the right eye and 20/40 in the left eye. Her CN III palsy had completely resolved on the right side. She was tapered to 10 mg/day of oral prednisone and was referred for follow-up with a pulmonologist for input regarding steroid-sparing medication, such as methotrexate, and for a repeat MRI. Through telephone correspondence, she reportedly was “living her life and getting around.”
|
Discussion
Although the patient’s past medical history skewed the differential diagnosis, other conditions that could present with similar imaging findings include tuberculosis; meningioma; other intracranial neoplasms; Rosai Dorfman disease (sinus histiocytosis with massive lymphadenopathy); and Churg-Strauss syndrome with granulomatous meningeal enhancement.
The brain is involved in known cases of systemic sarcoidosis in approximately 25 percent of cases, with only 20 percent of these cases demonstrating symptoms. In 2009, Siddharama Pawate, MBBS, and colleagues published a case series of 54 patients with neurosarcoidosis, demonstrating that the most common presentations of neurosarcoidosis were bilateral optic neuropathy (24 percent), myelopathy (19 percent), seizures (17 percent) and headache (17 percent). Neurosarcoidosis can also present with facial nerve palsy, hearing loss or other cranial nerve palsy, as in our patient. In this series, the most common MRI brain findings included T2 hyperintense, non-enhancing white matter lesions (30 percent), meningeal enhancement (19 percent), and contrast-enhancing parenchymal white matter lesions (19 percent). Interestingly, 72 percent of patients had an abnormal MRI of the spinal cord. This study was not performed in our patient, but would be something to be considered in the workup for neurosarcoidosis.
There does not appear to be a consensus on the medical management of neurosarcoidosis. Dr. Pawate and associates report a wide variety in the choice of medication for maintenance treatment after initial treatment with intravenous or oral steroids. This includes both steroid and steroid-sparing therapy. Twenty percent of patients had no specific treatment, and the remaining patients were divided between methotrexate, prednisone, mycophenolate mofetil, azathioprine, plaquenil, rituximab and combinations of these medications. Other researchers suggest a role for radiation therapy, but data is limited to case studies and small series. Unfortunately, despite treatment, the visual prognosis of neurosarcoidosis is poor, with more than 50 percent of patients with visual acuity of 20/200 or worse in at least one eye and nearly 25 percent with bilateral decreased vision from optic neuropathy, as reported by Dr. Pawate and colleagues. REVIEW
The author would like to acknowledge Jennifer Hall, MD, Wills Eye Neuro-Ophthalmology Service, and Jared Peterson, MD, for their time and assistance in preparing this case report.
1. Bruns F, Pruemer B, Haverkamp U, Fischedick AR. Neurosarcoidosis: An unusual indication for radiotherapy. Brit J Radiol 2004;77:777-9.
2. Johnson MD, Powell SZ, Boyer PJ, et al. Dural lesions mimicking meningiomas. Hum Pathol 2002;33:1211-26.
3.Kang S, Suh JH. Radiation therapy for neurosarcoidosis: Report of three cases from a single institution. Radiat Onc Investig,1999;7:309-12.
4. Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: A study of 54 cases. Q J Med 2009;102:449-60.
5. Raslan OA, Schellingerhout D, Fuller GN, Ketonen LM. Rosai-Dorfman disease in neuroradiology. Am J Roentgenol 2011;196:W187-93.
6. Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. Am J Roentgenol 2004; 182:289-95.
7. Tokumaru AM, Obata T, Kohyama S, Kaji T, et al. Intracranial meningeal involvement in Churg-Strauss syndrome. Am J Neuroradiol 2002;23:221-4.


