Workup, Diagnosis and Treatment
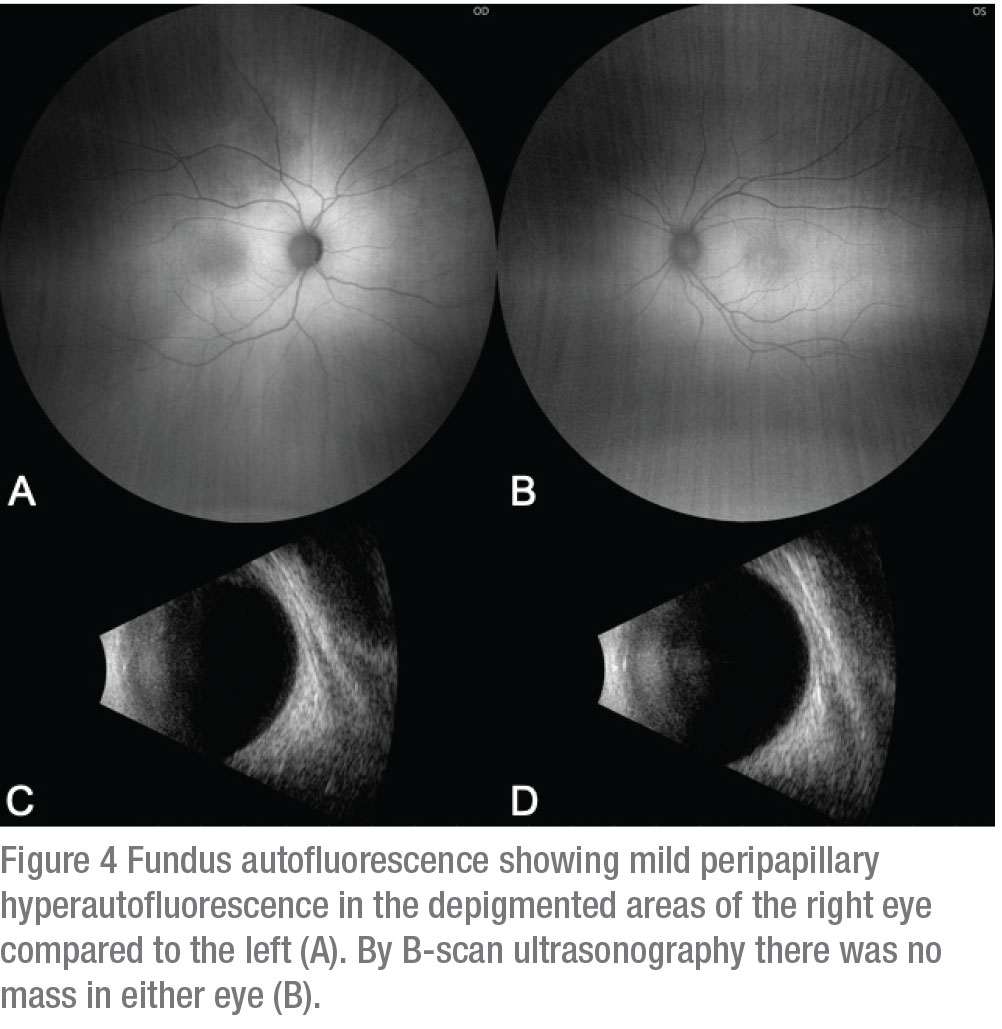
Further ancillary imaging was performed. Optical coherence tomography displayed normal foveal features OU, with subfoveal choroidal thickness at 210 µm OU, but the right eye demonstrated increased light transmission into deeper structures. Autofluorescence demonstrated mild scleral unmasking of autofluorescence in the right eye in the area of hypopigmentation compared to the left (Figure 4 A and B). By anterior segment OCT there was slight thinning of the iris stroma with loss of crypt architecture OD as compared to OS (Figure 1 C and D). There was no retinal or choroidal elevation on ultrasonography OU (Figure 4 C and D).
 |
On further questioning, the patient’s mother revealed that she had a white central portion of hair that she had routinely dyed since the age of 12 years. With the presence of iris hypopigmentation, choroidal hypopigmentation, synophrys, and a personal and family history of white forelock, the patient was diagnosed clinically with Waardenburg syndrome. As congenital deafness can be present in this condition, a complete auditory evaluation was performed and found to be normal.
Discussion
Waardenburg syndrome is a relatively rare congenital disease that occurs in one in 40,000 births.1 It was first described in 1951.2 This condition is composed of six defining features, including telecanthus or dystopia canthorum (displacement of the medial canthi), broad nasal root synophrys of the eyebrows, white forelock in the scalp, iris heterochromia and congenital sensorineural deafness.2 Dystopia canthorum is quantified using a “W Index,” which incorporates inner canthal distance, interpupillary distance and outer canthal distance. W Index values greater than 1.95 are considered abnormal.4 In 1966, researchers detailed the findings associated with WS and found a correlation between hypopigmentation of the iris and hypopigmentation of the choroid.3 Most recently, one of this report’s authors, Dr. Shields, and colleagues described these findings in detail and found both iris and choroid hypopigmented regions to be thinner on AS-OCT and posterior segment OCT, respectively.4 The hypopigmentation and thinning is believed to be related to abnormal neural crest migration and melanin production.5
Ocular melanocytosis is a close mimicker of WS, as the pigmentary abnormality can affect the uveal tract in both a sectoral and diffuse pattern.6 Unlike WS, melanocytosis is a hyperpigmentation and not a hypopigmentation. Additionally, ocular melanocytosis can manifest with increased pigmentation of the sclera, skin and palate. By AS-OCT, melanocytosis demonstrates iris thickening with loss of crypts and presence of mammillations from overabundant melanocytes, whereas WS shows thinning and loss of crypts. One further point is that WS can be hereditary and affect multiple family members, whereas melanocytosis isn’t hereditary. This distinction is important, as ocular melanocytosis is associated with secondary glaucoma and an increased likelihood of development of uveal melanoma, unlike WS.7
Four distinct types of WS have been described. WS1 and WS2 are inherited in an autosomal dominant fashion and are likely related to the originally described syndrome.4 There’s a large variability of phenotypic expression, and individuals will typically show only a few of the classical features. WS3 and WS4 seem to have an autosomal recessive pattern of inheritance. WS3 is associated with malformations of the upper limbs, and WS4 is associated with Hirschsprung disease.8 Multiple genes have been associated with WS, including EDN3, EDNRB, MITF, PAX3 and SOX10. The exact functions of these genes have yet to be elucidated.9 Genetic testing for individuals suspected to have WS may help confirm the diagnosis. Ultimately, patients with WS have a preserved visual prognosis and require annual ophthalmic examination. It’s important to remember that Waardenburg Syndrome accounts for 2 to 5 percent of all patients with congenital hearing loss.1
In conclusion, in patients who present with diffuse choroidal hyper- or hypopigmentation in both eyes, consider that the lighter areas may represent WS, rather than the dark areas of ocular melanocytosis, a close mimicker. Examination for classical features of WS including telecanthus (dystopia canthorum), synophrys of the eyebrows, white forelock, broad nasal root, heterochromia and congenital deafness can lead to the diagnosis. And don’t forget that a detailed family history—including asking questions about the dyeing of hair—can be revealing, as in this case. REVIEW
1. Zaman A, Capper R, Daddoo W. Waardenburg syndrome: More common than you think! Clin Otolaryngol 2015;40:1:44-8.
2. Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet 1951;3:3:195-253.
3. Goldberg MF. Waardenburg’s syndrome with fundus and other anomalies. Arch Ophthalmol 1966;76:6:797-810.
4. Shields CL, Nickerson SJ, Al-Dahmash S, Shields JA. Waardenburg syndrome: Iris and choroidal hypopigmentation: Findings on anterior and posterior segment Imaging. JAMA Ophthalmol 2013;131:9:1167–1173.
5. Mullaney PB, Parsons MA, Weatherhead RG, Karcioglu ZA. Clinical and morphological features of Waardenburg syndrome type II. Eye (Lond) 1998;12:3a:353-357.
6. Shields CL, Qureshi A, Mashayekhi A,et al. Sector (partial) oculo(dermal) melanocytosis in 89 eyes. Ophthalmology 2011;118:12:2474-2479.
7. Singh AD, De Potter P, Fijal BA, et al. Lifetime prevalence of uveal melanoma in Caucasian patients with oculo(dermal) melanocytosis. Ophthalmology 1998;105:195–8.
8. Pingault V, Ente D, Dastot-Le Moal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat 2010;31:4:391-406.
9. Tachibana M. A cascade of genes related to Waardenburg syndrome. J Investig Dermatol Symp Proc 1999;4:2:126-29.


