Presentation
A 72-year-old Caucasian male was referred to the Ocular Oncology Service at Wills Eye Hospital for a plaque-like lesion on the right upper eyelid. He had noticed thickening of the right upper eyelid and development of a hard nodule one year prior. An incisional biopsy by the referring oculoplastic surgeon revealed an atypical vascular neoplasm. The patient had no cervical lymphadenopathy and was otherwise asymptomatic.
History
Past ocular history was unremarkable aside from previous cataract surgery in both eyes. Past medical history was notable for chronic obstructive pulmonary disease and benign prostatic hyperplasia. He had previous surgical replacement of both hip joints and lumbar spinal fusion. The patient had no history of cancer, including skin cancer. Family history included liver cancer in his father, macular degeneration in his mother and systemic lupus erythematosus in his sister. Social history was significant for previous tobacco use, though he quit smoking more than 50 years ago. There was no social or occupational history of extensive ultraviolet light or radiation exposure. Current medications included fluticasone-salmeterol for chronic obstructive pulmonary disease and finasteride for benign prostatic hyperplasia; he had no known drug allergies.
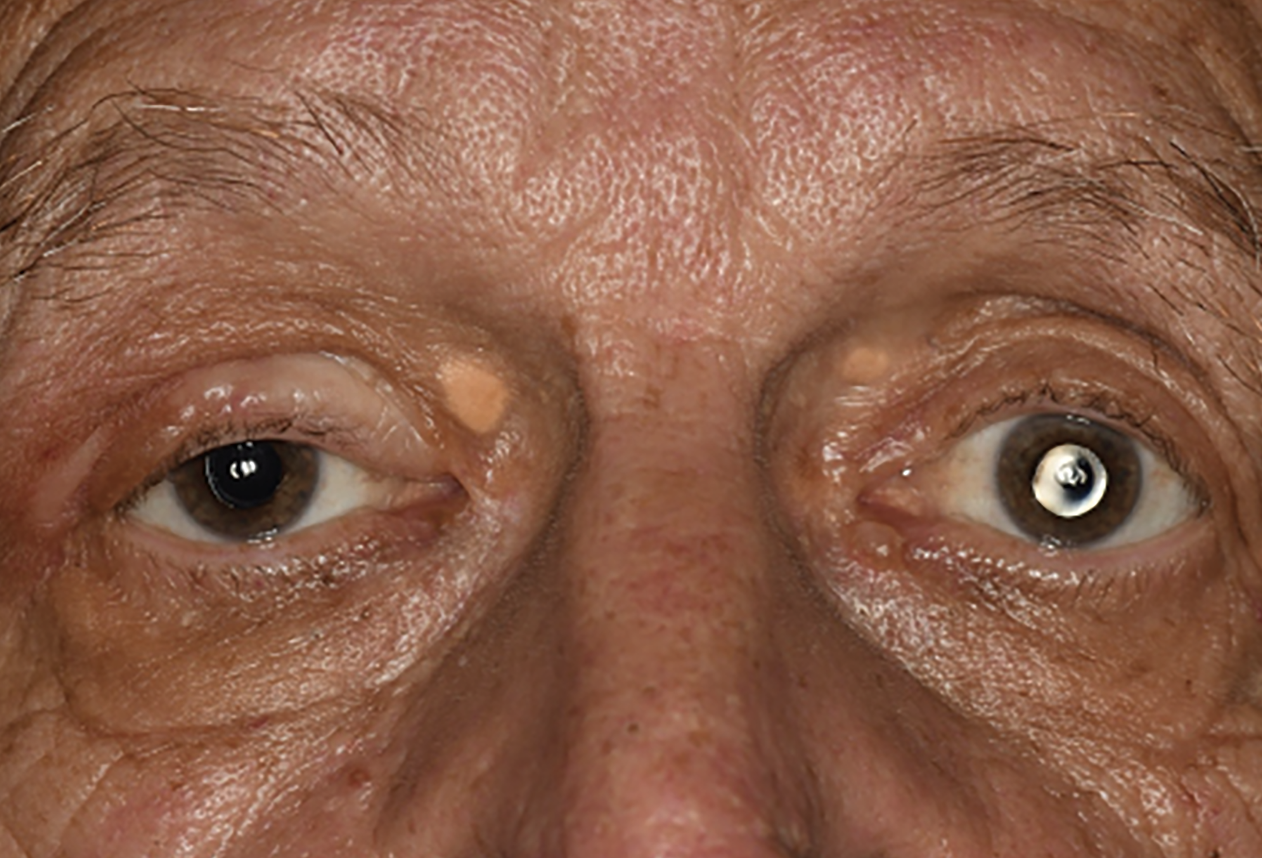 |
| Figure 1. External photograph demonstrates subtle right upper eyelid mass with edema, a healed scar of prior biopsy and blepharoptosis. Bilateral xanthelasmas are also present superonasally. |
Examination
Visual acuity was 20/20 in the right eye and 20/40 in the left eye with pinhole improvement to 20/30. Pupils were round and reactive to light with no relative afferent pupillary defect. Intraocular pressure was 14 mmHg in the right and 11 mmHg in the left eye. Examination of the eyelids and adnexa demonstrated a 40 x 30 mm, diffuse, subtly violaceous thickening of the right upper eyelid with a well-healed overlying incision (Figure 1). Lashes were intact. There was a 10 x 10 mm palpable nodule superior to the right lateral canthus and bilateral medial eyelid xanthelasma. Anterior segment examination was notable only for bilateral posterior capsule intraocular lenses in good position, and dilated ophthalmoscopic examination was unremarkable.
What’s your diagnosis? What further work-up would you pursue? The diagnosis appears below.
Work-up, Diagnosis and Treatment
Pathology review of prior biopsy and discussion at the multidisciplinary tumor board led to a working differential of low-grade angiosarcoma vs. possible benign and borderline vascular neoplasms, such as hemangioma and hemangioendothelioma. Magnetic resonance imaging of the orbits showed a thickened enhanced right upper eyelid, without a discrete mass or lymphadenopathy. Six weeks after initial referral to ocular oncology, the patient was taken to the operating room for incisional biopsy. Histopathologic examination showed an infiltrating neoplasm composed of vascular channels lined by mildly pleomorphic spindle and ovoid cells. Mitotic figures were not conspicuous. There was no evidence of necrosis (Figure 2). Neoplastic cells co-expressed vascular and lymphatic endothelial markers erythroblast transformation-specific [ETS]-related gene (ERG) and CD31, and lymphatic endothelial marker D2-40 (Figure 3A-C). Neoplastic cells were negative for human herpes virus-8 (HHV8), excluding the diagnosis of Kaposi sarcoma. The Ki-67 proliferative index was low (Figure 3D). Although there were no high-grade morphologic features typical of angiosarcoma, the infiltrative pattern of this vascular proliferation was concerning for well-differentiated angiosarcoma.
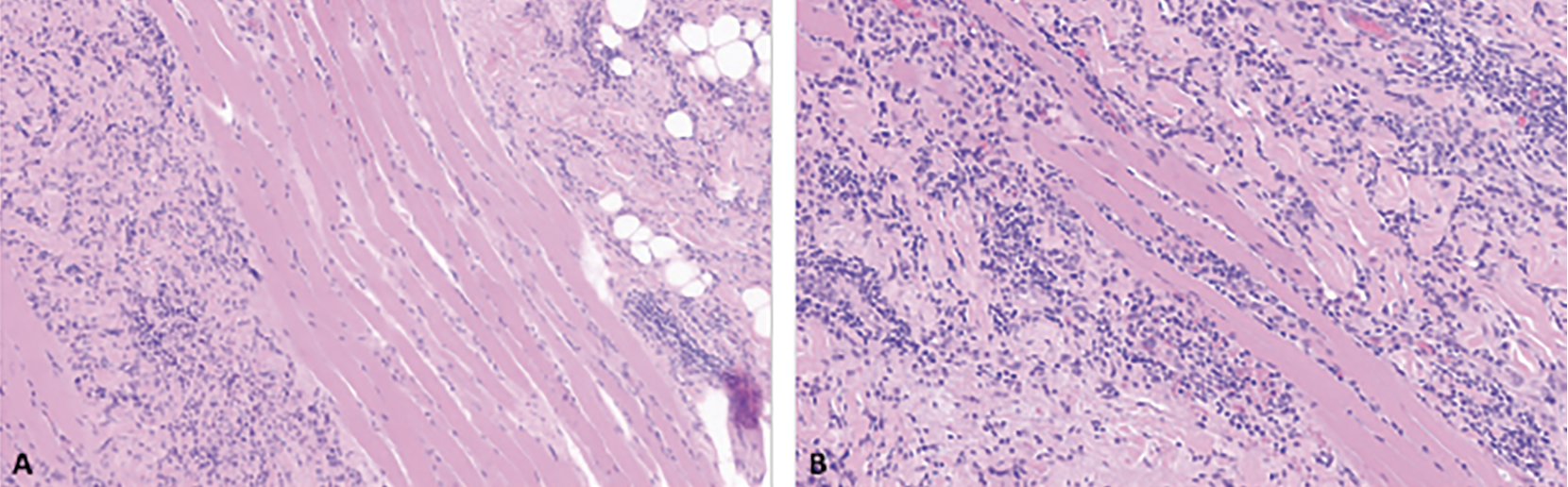 |
| Figure 2. Primary eyelid angiosarcoma, initial resection, histologic findings. Infiltrative neoplasm involving adipose tissue (A) and skeletal muscle (B) composed of vascular channels lined by mildly pleomorphic spindle and ovoid cells (hematoxylin-eosin stain; 100x (A), 150x (B)). |
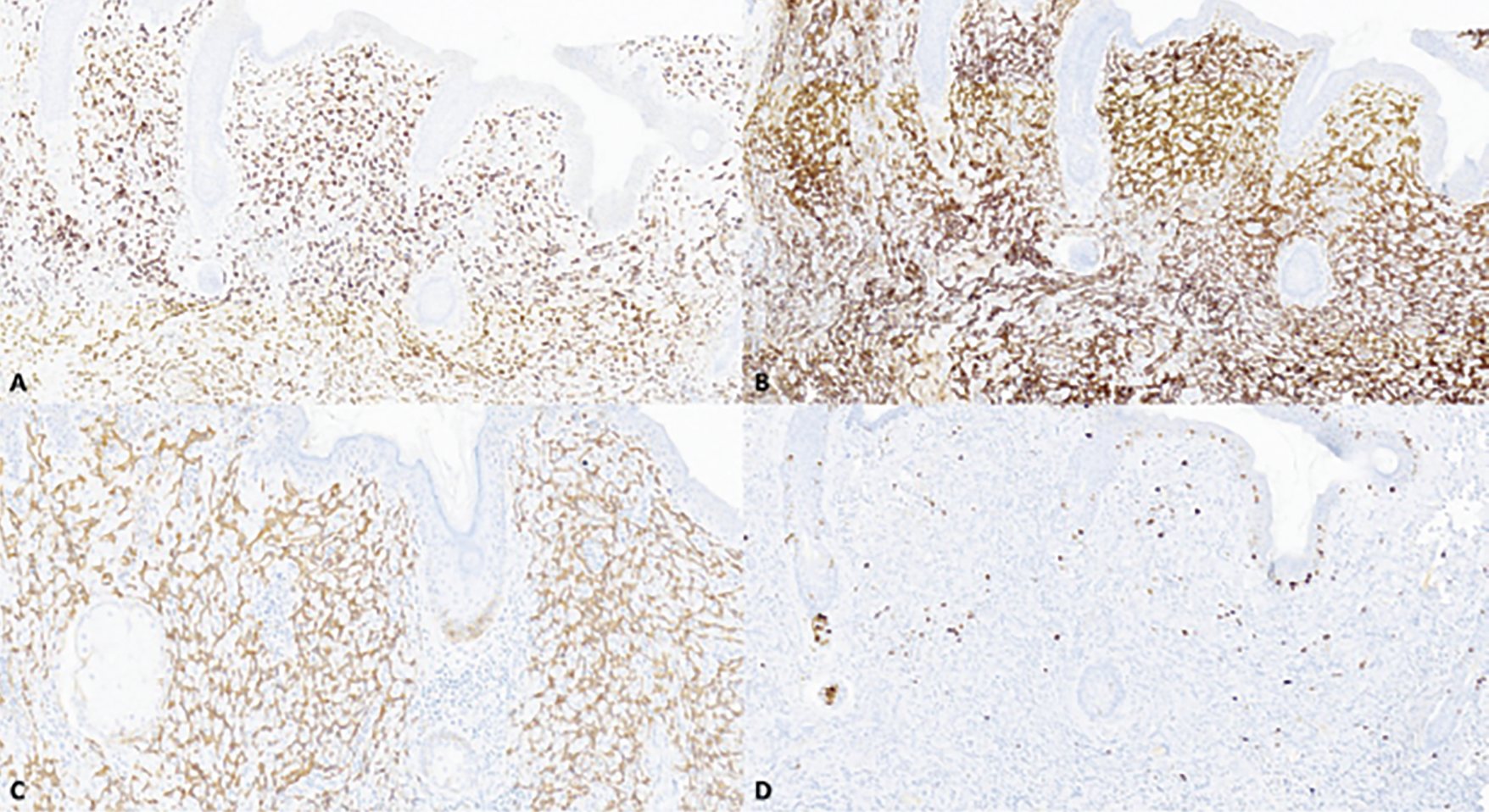 |
| Figure 3. Primary eyelid angiosarcoma, initial resection, immunohistochemical findings. The neoplastic cells express erythroblast transformation-specific [ETS]-related gene [ERG] (A), CD31 (B) and D2-40 (C). The Ki-67 proliferative index is low (<1%) (D). All images are 100x magnification. |
After discussion at the orbital tumor board, the decision was made to observe the patient closely given the low-grade morphology and low risk for metastatic disease.
Six months later, repeat MRI demonstrated interval increased enhancement of the right upper eyelid and no orbital or lacrimal mass. The patient underwent repeat biopsy 11 months after initial presentation to ocular oncology, which showed a neoplasm morphologically and immunohistochemically similar to the initial biopsies. Because of the locally aggressive behavior of the tumor, the patient underwent systemic neoadjuvant chemotherapy with taxol prior to a definitive tumor resection. Histopathologic evaluation of the resected tumor showed an infiltrative vascular neoplasm with a more complex architecture when compared to the initial biopsies, supporting the diagnosis of well-differentiated angiosarcoma (Figure 4).
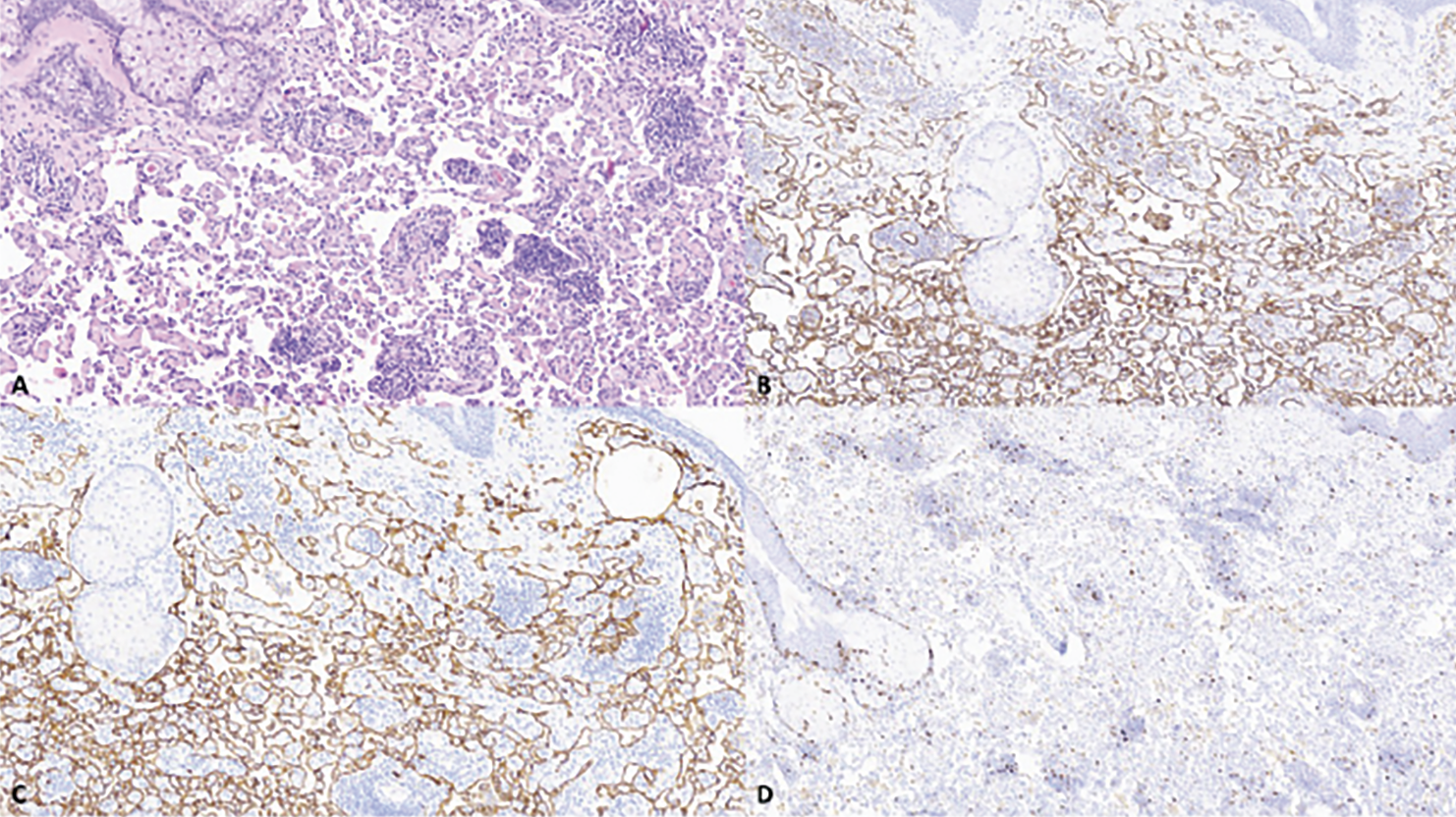 |
| Figure 4. Primary eyelid angiosarcoma, recurrent lesion. Recurrent lesion has a more complex architecture when compared to the initial resection (A). The complex vascular network is highlighted with CD31 (B) and D2-40 (C). Low Ki-67 proliferative index (<1%) (D). All images are 100x magnification. |
Discussion
Primary angiosarcoma of the eyelid is a rare and aggressive sarcoma that shows vascular and lymphatic endothelial cell differentiation. Ultraviolet light exposure and radiation exposure are well-recognized risk factors for angiosarcoma of the head and neck skin, including the eyelid.1 Diagnosis of eyelid angiosarcoma can be difficult because the presenting clinical features are diverse and may be subtle. In a systematic review of 42 eyelid angiosarcoma cases, this neoplasm was most commonly described as having reddish, erythematous color and less frequently has been reported to appear violaceous, yellow, brown or without any change in color.2 The lesion shape was frequently described as a papular or plaque-like mass or edema of the eyelid. However, there were also cases of color change without any apparent induration or tumefaction.3 As such, eyelid angiosarcoma may be a difficult diagnosis to make clinically and is frequently misdiagnosed prior to biopsy.4,5 The most frequent preoperative diagnoses for primary eyelid angiosarcoma are cellulitis (19 percent), angioedema (12 percent) and hematoma (5 percent).2 Therefore, for erythematous eyelid swelling or mass-like lesions that don’t appear to resolve with initial anti-inflammatory therapy, clinicians may consider biopsy to exclude this malignancy.
Histopathologic diagnosis of eyelid angiosarcoma can be particularly challenging. Angiosarcomas are usually high-grade neoplasms. However, eyelid angiosarcoma shows a spectrum of histopathologic findings, ranging from well-differentiated to poorly-differentiated tumors.4,5 Well-differentiated angiosarcomas are characterized by endothelial cells with mild nuclear atypia and few mitotic figures, similar to benign vascular lesions such as hemangioma and pyogenic granuloma. In these cases, an infiltrative growth pattern should raise your suspicion of malignancy, but the diagnosis is challenging in a small biopsy as exemplified by our patient’s tumor. Poorly differentiated angiosarcomas are typically composed of markedly atypical cells, frequently with epithelioid morphology and markedly pleomorphic nuclei in complex arrangements or in sheets, raising consideration of poorly differentiated carcinoma and other sarcomas. Immunohistochemical staining can help in these challenging cases. Angiosarcoma frequently co-expresses vascular and lymphatic endothelial markers (ERG, CD31, and D2-40) and is negative for HHV8, which excludes Kaposi sarcoma.
The four-year overall survival of patients with eyelid angiosarcoma was 49 percent in a review of previously published cases.2 Patients with angiosarcoma isolated to the eyelid had 100-percent survival, and those with diffuse disease involving adjacent facial structures had 57-percent survival at three years.3 The data on the prognostic value of tumor differentiation in periocular angiosarcoma are mixed and limited.6 Some studies suggest that poorly differentiated tumors with epithelioid morphology and necrosis are associated with increased mortality.3,7,8 Treatment varies by location and characteristics of the angiosarcoma, but typically involves a combination of surgical resection, chemotherapy and radiotherapy. The rarity of periocular angiosarcoma means there is little data to guide formal recommendations for treatment. Surgical resection of eyelid angiosarcoma is associated with significantly decreased mortality and recurrence, compared to patients who receive chemotherapy, radiotherapy, or combined chemotherapy and radiotherapy.2 However, complete resection is often challenging because of the tendency to widely infiltrate along the skin and other periocular tissues. Another analysis of cutaneous angiosarcoma involving the periorbita reported a 60-percent recurrence among patients who underwent neoadjuvant chemotherapy alone and a 40-percent recurrence in patients who underwent surgical resection with adjuvant chemotherapy or radiotherapy.9 As such, patients with lid angiosarcoma require close follow-up to monitor for recurrence, regardless of treatment.
This case of primary eyelid angiosarcoma is unusual because angiosarcoma typically involves the scalp, forehead, and face and may secondarily extend into the eyelid. However, primary eyelid angiosarcoma is very rare, with fewer than 50 patients reported in the literature. The diagnosis is challenging because eyelid angiosarcoma can mimic the features of many other benign and malignant eyelid lesions.
1. Painter CA, Jain E, Tomson BN, et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nature Medicine 2020;26:2:181-187.
2. Hwang G, Shin J, Lee JY, et al. The eyelid angiosarcoma: A systematic review of characteristics and clinical course. J Clin Med 2022;11:14.
3. Papalas JA, Manavi CK, Woodward JA, Sangueza OP, Cummings TJ. Angiosarcoma of the eyelid: A clinicopathologic comparison between isolated unilateral tumors and tumors demonstrating extrapalpebral involvement. Am J Dermatopathol 2010;32:7:694-699.
4. Ferguson DC, Mawn LA, Al-Rohil RN. Cutaneous angiosarcoma of the eyelid mimicking morbihan disease. Am J Dermatopathol 2018;40:8:617-620.
5. Milman T, Shields CL, Brooks JSJ, et al. Primary cutaneous angiosarcoma of the eyelid: A diagnostic and therapeutic challenge. Ocul Oncol Pathol 2018;4:4:230-235.
6. Shustef E, Kazlouskaya V, Prieto VG, Ivan D, Aung PP. Cutaneous angiosarcoma: A current update. J Clin Pathol 2017;70:11:917-925.
7. Dettenborn T, Wermker K, Schulze HJ, Klein M, Schwipper V, Hallermann C. Prognostic features in angiosarcoma of the head and neck: A retrospective monocenter study. J Craniomaxillofac Surg 2014;42:8:1623-1628.
8. Deyrup AT, McKenney JK, Tighiouart M, Folpe AL, Weiss SW. Sporadic cutaneous angiosarcomas: A proposal for risk stratification based on 69 cases. Am J Surg Pathol 2008;32:1:72-77.
9. DeMartelaere SL, Roberts D, Burgess MA, et al. Neoadjuvant chemotherapy-specific and overall treatment outcomes in patients with cutaneous angiosarcoma of the face with periorbital involvement. Head & Neck 2008;30:5:639-646.


