T. Cunningham Jr., MD, PhD, MPH,
Coats' disease is a form of idiopathic retinal telangiectasia most often occurring unilaterally in male children and young adults.Patients present with decreased visual acuity, leukocoria or strabismus. Peripheral retinal telangiectasias consisting of dilated capillaries and small aneurysms lead to the accumulation of fluid and exudate both intraretinally and subretinally in the posterior pole. This process may eventually lead to exudative retinal detachment and profound vision loss.
In 1908, George Coats described three subtypes of retinal disease characterized by massive retinal exudation and varying degrees of vascular abnormalities.1 He subsequently revised his classification after concluding that two of the three subtypes were manifestations of a single clinical condition (the third was recognized as a separate entity previously described by Eugen von Hippel).2 Recent literature has sought to clarify the definition of Coats' disease, specifying that the term "Coats' disease" should refer only to idiopathic congenital retinal telangiectasia with intraretinal or subretinal exudation and without appreciable vitreoretinal traction. This strict definition has been applied to distinguish true Coats' disease from the myriad of conditions that result in either retinal telangiectasia or retinal exudation. The term "Coats-like reaction" has been applied to the exudation that occurs with conditions such as retinitis pigmentosa. Unlike Coats' disease, this type of exudation is often bilateral and results from a loss of vascular integrity that may be directly attributed to the underlying condition.3 There is no known association between true Coats' disease and any systemic abnormalities.
Demographics
The vast majority of Coats' disease is diagnosed in the first decade of life, although mild forms of the condition can present well into adulthood.4,5 The disease occurs sporadically, and no hereditary pattern has been identified despite reports that individual cases of Coats' disease have occurred as a result of localized mutations in proteins governing angiogenesis.6 More than 75 percent of patients are male, and 95 percent of cases are unilateral.4 There is no clear racial predilection. The occurrence of retinal telangiectasis and exudation bilaterally, in female patients, or in adults should prompt a search for non-Coats causes of these findings.
Presentation and Clinical Findings
The most common presenting signs of Coats' disease are decreased visual acuity, strabismus and leukocoria. The visual acuity is highly variable, but the majority of cases have a presenting visual acuity of 20/200 or worse, and the presenting visual acuity appears to correlate strongly with long-term prognosis.4,7 Examination rarely reveals anterior segment findings, although severe cases may show corneal edema, anterior chamber shallowing, suspension of lipid and protein in the aqueous humor, or neovascular glaucoma.
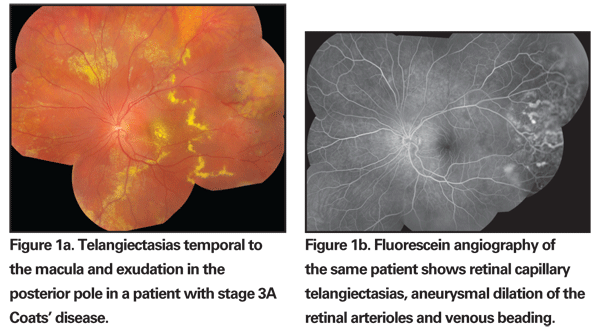
Posterior segment findings range from mild peripheral telangiectasia of the retinal vasculature to florid exudation with total retinal detachment. The telangiectatic vessels are rarely located in the macula, and more than half the telangiectasias are found anterior to the equator. For unclear reasons, the telangiectatic vessels appear to have a predilection for the temporal and inferior quadrants.4 The telangiectasias may be accompanied by grape-like clusters of microaneurysms, fusiform dilation of retinal arterioles ("light bulb aneurysms"), sheathing of retinal vessels, or venous beading (See Figures 1a & b).
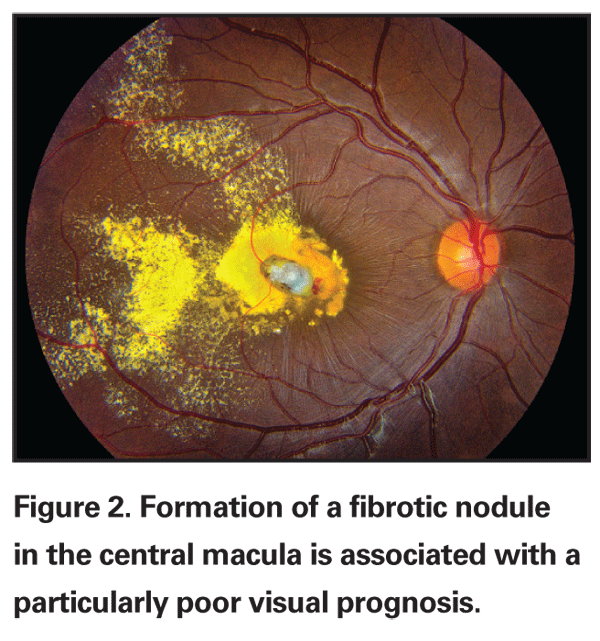
Despite the peripheral location of the telangiectasias, the intraretinal and subretinal exudates often migrate toward the macula, leading to severe visual loss despite the large distance between the primary pathology and the fovea. The reason for this macular accumulation of exudates is not known, but it has been hypothesized that the retinal pigment epithelium in this area pumps more actively, acting as a sump within the eye and drawing the fluid and exudates into the posterior pole. The exudates that accumulate in Coats' disease contain unusually high levels of cholesterol and protein, and these components are concentrated in the macula as free water is extracted by the retinal pigment epithelium.9 In cases of massive exudation, a dense nodule may form in the macula, which is associated with a particularly poor prognosis (See Figure 2). Histologic studies indicate that the composition of the nodule is highly cellular, consisting of lipid and hemosiderin-laden macrophages and retinal pigment epithelial cells with fibrous metaplasia, but the mechanism for formation of the nodule is not fully understood.10-12
Patients typically present with varying degrees of exudative retinal detachment, and the majority of patients will develop a significant retinal detachment if left untreated. The detachment usually begins in the area of the telangiectasia, subsequently involving the macula as a result of exudation, and eventually progressing to total retinal detachment. Advanced cases may develop a secondary glaucoma as a result of forward displacement of the lens-iris diaphragm or neovascularization of the iridocorneal angle.3
Classification
In 2000, a classification scheme for Coats' disease was proposed that correlates the stages of the disease with known indicators of long-term prognosis.13 This system utilizes five stages, representing the spectrum from isolated telangiectasia to end-stage neovascular glaucoma with phthisis. Stage 1 disease consists of telangiectasia only; stage 2 indicates that both telangiectasia and exudation are present (2A, extrafoveal exudation; 2B, foveal exudation); stage 3 indicates retinal detachment (3A, subtotal retinal detachment; 3B, total retinal detachment); stage 4 consists of total retinal detachment and secondary glaucoma; and stage 5 indicates end-stage disease approaching phthisis. Poor visual outcome (20/200 or worse) is rare in stage 1 disease, but occurs in half of patients with stage 2 disease and nearly 75 percent of those with stage 3 disease, even when those patients receive aggressive treatment. Patients with stage 4 or stage 5 disease invariably have a poor visual prognosis.13
Ancillary Testing
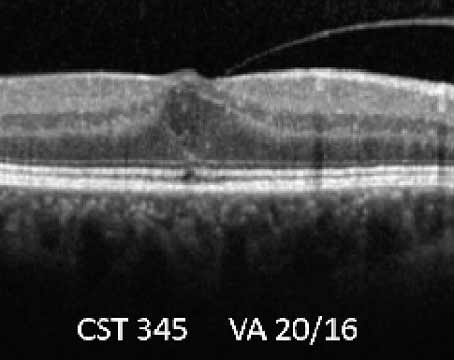
The diagnosis of Coats' disease is primarily based on clinical characteristics alone and examination under anesthesia is often required in children who are unable to tolerate a careful funduscopic examination. Ancillary testing is sometimes helpful in determining the severity of the disease, or in differentiating it from other causes of leukocoria and vision loss in children. Fluorescein angiography will highlight areas of telangiectasia, and may also show microaneurysms, areas of capillary nonperfusion, or retinal neovascularization. Areas of anomalous vasculature will leak in the late phases of the angiogram.14 When the diagnosis is unclear, ultrasound or CT scan may be needed to rule out retinal detachment secondary to a mass lesion, and are particularly useful for identifying the calcifications of retinoblastoma. Optical coherence tomography can be used to evaluate the extent of intraretinal and subretinal fluid, exudates or fibrosis in the macula.
Differential Diagnosis
The differential diagnosis of Coats' disease varies based on the stage at presentation. Advanced Coats' disease must be differentiated from other causes of non-rhegmatogenous retinal detachment including: retinoblastoma; persistent fetal vasculature; retinopathy of prematurity; familial exudative vitreoretinopathy; retinal capillary hemangioma; retinal cavernous hemangioma; incontinentia pigmenti; Norrie's disease; and toxocariasis. A "Coats-like reaction" consisting of vascular abnormalities and varying degrees of exudation has been described in retinitis pigmentosa,15 branch retinal vein occlusion,16 toxoplasmosis17 and systemic conditions such as muscular dystrophy,18,19 Turner syndrome,20 Alport syndrome21 and others.
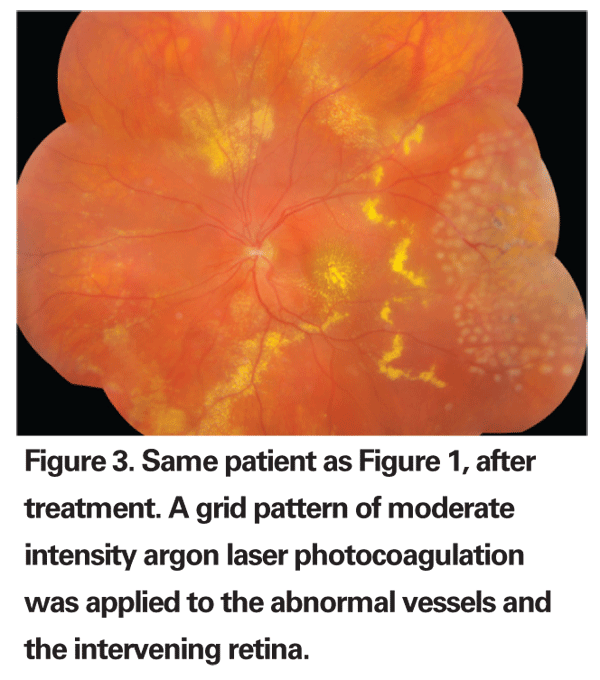
Treatment
Treatment options for Coats' disease are best tailored to disease severity. Observation may be appropriate in some patients with stage 1 Coats' disease (telangiectasia without exudates), but treatment should generally be applied at the first signs of exudation, given that stage 2 disease will often progress if untreated. Scatter laser photocoagulation to the area of telangiectasia with direct treatment to the abnormal vessels and moderately intense retinal burns in between is preferred if the retina is attached (See Figure 3). For stage 3 disease, cryotherapy is useful for treating the detached retina. Overly aggressive treatment may worsen the retinal detachment, and repeat treatments should generally be spaced at least three months apart. This also allows adequate time for the resorption of subretinal fluid and provides a more accurate assessment of treatment efficacy.13
Subfoveal fluid and macular exudation have been seen to worsen after laser or cryotherapy in some patients, and it has been suggested that this may be reduced or prevented by placing several rows of barrier laser posterior to the area of telangiectatic vessels in a region of attached retina. This barrier is intended to prevent posterior migration of fluid after subsequent treatment of the telangiectasias themselves.7 Patients with extensive retinal detachment may benefit from scleral buckling, vitrectomy or both.22 Patients with stage 4 or stage 5 disease, particularly those with neovascular glaucoma or phthisis bulbi, may require enucleation.
Prognosis
The primary factors determining final visual outcome in patients with Coats' disease are the presenting visual acuity, extent of retinal detachment and the extent of macular exudation. As previously mentioned, cases with florid macular exudation may develop subfoveal fibrosis, which is almost invariably associated with a poor visual prognosis.3 Stage 1 disease rarely results in severe visual loss, but stage 2 disease will progress to a final acuity of 20/200 or worse in half of patients, even with aggressive treatment.
Treatment in stage 3 and 4 disease is primarily aimed at anatomic correction of the retinal detachment, lowering of intraocular pressure and prevention of phthisis. Stabilization or improvement of visual acuity is generally seen in patients with less than four clock hours of involved retina.7 It has been noted that visual outcomes are particularly poor in Coats patients referred with an initial diagnosis of retinoblastoma, presumably due to the extent of retinal detachment.23 Long-term follow-up of patients with Coats' disease has shown that the condition can recur after long periods of quiescence, and it should be emphasized that periodic examination is important even in patients who have been stable for decades.24
A century after it was originally described, Coats' disease remains a cause of severe vision loss in the young. Slit-lamp examination and indirect ophthalmoscopy alone can often differentiate this disease from other causes of retinal detachment. When necessary, ultrasonography, fluorescein angiography and computed tomography can assist in making the diagnosis and ruling out other disorders such as retinoblastoma.
While we now enjoy greater diagnostic accuracy and are able to salvage more eyes with Coats' disease, the rates of central vision loss remain high due to complications such as retinal detachment, macular fibrosis and amblyopia. Future research in this dis-ease should focus on earlier detection and methods to maintain macular function.
Dr. Pomerleau is a resident in ophthalmology at
1. Coats G. Forms of retinal disease with massive exudation. Roy Lond Ophth Hosp Rep 1908; 17:440-525.
2. Coats G. Ueber retinitis exudativa (retinitis haemorrhagica externa) Graefe's Arch Ophthal 1912, 81:275-327.
3. Shields JA, Shields CL. Review: Coats' disease: The 2001 LuEsther T. Mertz lecture. Retina 2002;22:80-91.
4. Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats' disease in 150 cases: The 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001;131:561-571.
5. Smithen LM, Brown GC, Brucker AJ, Yannuzzi LA, et al. Coats' disease diagnosed in adulthood. Ophthalmology 2005;112(6):1072-1078.
6. Black GC, Perveen R, Bonshek R, Cahill M, Clayton-Smith J, Lloyd IC, McLeod D. Coats' disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: A role for norrin in retinal angiogenesis. Hum Mol Genet 1999;8(11):2031-2035.
7. Budning AS, Heon E, Gallie BL. Visual prognosis of Coats' disease. J AAPOS 1998;2(6):356-359.
8. Gass JDM. Stereoscopic atlas of macular diseases; a funduscopic and angiographic presentation.
9. Hsu J, Forbes B, Maguire AM. Total exudative retinal detachment in Coats' disease: Biochemical analysis of the subretinal exudate. Retina 2006;26:831-833.
10. Jonas JB, Holbach LM. Clinical-pathologic correlation in Coats' disease. Graefe's Arch Clin Exp Ophthalmol 2001;239(7):544-545.
11. Chang MM, McLean IW, Merritt JC. Coats' disease: A study of 62 histologically confirmed cases. J Pediatr Ophthalmol Strabismus 1984;21(5):163-168.
12. Khurana RN, Samuel MA, Murphree AL, Loo RH, Tawansy KA. Subfoveal nodule in Coats' disease. Clin Experiment Ophthalmol 2005;33(3):301-302.
13. Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats' disease: The 2000 Proctor Lecture. Am J Ophthalmol 2001;131:572-583.
14. Jones JH, Kroll AJ,
15. Khan JA, Ide CH, Strickland MP. Coats'-type retinitis pigmentosa. Surv Ophthalmol 1988;32(5):317-332.
16. Luckie AP,
17. Frezzotti R, Berengo A, Guerra R, Cavalllini F. Toxoplasmic Coats' Retinitis. A Parasitologically Proved Case. Am J Ophthalmol 1965;59:1099-1102.
18. Desai UR, Sabates FN. Long-term follow-up of facioscapulohumeral muscular dystrophy and Coats' disease. Am J Ophthalmol 1990;110:568-569.
19. Shields CL, Zahler J, Falk N, Furuta M, et al. Neovascular glaucoma from advanced Coats' disease as the initial manifestation of facioscapulohumeral dystrophy in a 2-year-old child. Arch Ophthalmol 2007;125:840-842.
20. Cameron JD, Yanoff M, Frayer WC. Coats' disease and turner's syndrome. Am J Ophthalmol 1974;78:852-854.
21. Kondra L, Cangemi FE, Pitta CG. Alport's syndrome and retinal telangiectasia. Ann Ophthalmol 1983;15(6):550-551.
22. Yoshizumi MO, Kreiger AE, Lewis H, Foxman B, Hakakha BA. Vitrectomy techniques in late-stage Coats'-like exudative retinal detachment. Doc Ophthalmol 1995;90(4):387-394.
23. Char DH. Coats' syndrome: Long term follow up. Br J Ophthalmol 2000;84(1):37-39.
24. Shienbaum G, Tasman WS. Coats' disease: A lifetime disease. Retina 2006;26:422-424.