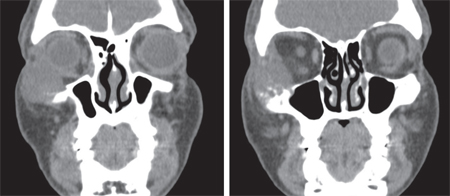
Based on the clinical history and exam, the differential diagnosis included inflammatory or reactive conditions such as a hematoma; aneurysmal bone cyst; giant cell or cholesterol granuloma; histiocytosis; and idiopathic orbital inflammation. The differential diagnosis also included infectious and neoplastic disorders such as cellulitis, primary bone lesions (benign or malignant), lymphoproliferative or vascular disorders, and secondary neoplasms including sinus-related lesions or distant metastases.1,2
|
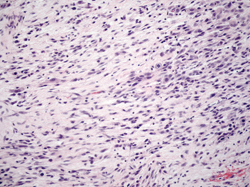
The patient underwent an orbital biopsy. Resulting pathology showed sheets of numerous strap cells with abundant eosinophilic cytoplasm reminiscent of rhabdomyoblasts and scattered mitotic figures (See Figure 3). Immunohistochemistry stains were positive for desmin, myogenin, AE1/AE3 and CAM 5.2.
The patient was diagnosed with orbital spindle cell rhabdomyosarcoma and was referred to the Wills Eye Hospital Ocular Oncology service as well as the Thomas Jefferson University Hospital Otolaryngology and Hematology/Oncology departments. A PET/CT showed that disease was localized to the right orbit. While all options, including observation, chemotherapy, radiotherapy and surgical debulking/resection were discussed, the final recommendation was to pursue a combination of systemic and localized therapy in the form of neoadjuvant chemotherapy (vincristine, adriamycin, and cyclophosphamide) followed by local radiation ±local debulking. Unfortunately, after only one visit in each of the previously mentioned departments, the patient refused further therapy and failed to return for any further visits.
Discussion
Rhabdomyosarcoma is a neoplasm that develops from undifferentiated mesenchymal cells that have the capacity to differentiate into striated muscle. Based on the widely adopted generalized Horn and Enterline classification system, there are classically four distinct histopathological subtypes, each with its own characteristic and identifying features: embryonal; botyroid variant of embryonal; alveolar; and pleomorphic. Although used in the original Intergroup Rhabdomyosarcoma Studies, in recent years this scheme has been modified and adapted by investigating organizations including the National Cancer Institute with the goal of creating a classification system that would better predict patient outcome.3
|
Orbital rhabdomyosarcoma is most commonly a disease of pediatric patients who usually present with symptoms before the age of 10 years. It represents approximately 4 percent of pediatric malignancies and is the most common soft-tissue sarcoma of the head and neck in this patient population. Initial presenting symptoms usually include rapidly developing proptosis, globe displacement, eyelid edema, and ptosis. In contrast, adult cases such as ours are much more rare.
|
In terms of staging, categories are classically defined based on the Intergroup Rhabdomyosarcoma Study post-biopsy system (See Table 1).11 Favorable prognostic factors include an orbital location, younger age (1 to 10 years), female sex, embryonal histology, and low tumor burden (size <5 cm diameter).4-6,12
Treatment approaches usually correlate with the stage of disease. Specifically, patients with stage I disease usually receive chemotherapy with either vincristine or actinomycin. For those with stage II or III disease, some combination of chemotherapy (typical agents being vincristine, actinomycin and cyclophoshamide) and radiotherapy is the standard. Finally, for those with metastatic or stage IV involvement, an intensive chemotherapy and radiation regimen is used, along with intrathecal chemotherapy for patients with intracranial involvement. With advances in chemotherapy and radiotherapy over the past 50 years, surgical treatment is no longer the standard of care, and survival rates have markedly improved from 30 percent to 90 percent. In fact, while surgical excision and exenteration used to be the primary treatment, these approaches are often reserved for cases with recurrent disease.4,11,13
In summary, rhabdomyosarcoma is the most common primary orbital and soft tissue malignancy in children, but can occur in patients of any age. Four distinct histopathological subtypes have different frequency rates and prognoses. Fortunately, due to advances in chemotherapy and radiotherapy over the past fifty years, survival rates have markedly improved and disfiguring procedures such as exenteration are no longer the standard of care. REVIEW
The author would like to thank Robert Penne, MD, and Michael Rabinowitz, MD, members of the Wills Eye Hospital Oculoplastic and Orbital Surgery Service, for their time and assistance in preparing this case report.
1. Heggeness MH, et al. Orthopedic Surgery. In: Brunicardi FC, ed. Schwartz’s Principles of Surgery. 9th ed. New York, NY: McGraw-Hill, 2010.
2. Srinivasan RC, Tolhurst SR, Vanderhave K. Orthopaedics. In: Doherty GM (ed): Current Surgical Diagnosis and Treatment, ed. 13. New York, NY: McGraw-Hill Medical, 2009:1006-1091.
3. Newton, WA, et al. Classification of rhabdomyosarcomas and related sarcomas. Cancer 1995;76(6):1073-85.
4. Jurdy L, et al. Orbital rhabdomyosarcomas: A review. Saudi J Ophthalmol 2013;27:167-75.
5. Karcioglu ZA, et al. Orbital rhabdomyosarcoma. Cancer Control 2004;11.5:328-33.
6. Shields CL, et al. Primary ophthalmic rhabdomyosarcoma in 33 patients. Trans Am Ophthalmol Soc 2001;99:133-144.
7. Sorensen PHB, et al. PAX3-FKHR and PAX7-FKHR Gene Fusions Are Prognostic Indicators in Alveolar Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J Clinical Oncology 2002; 20(11):2672-79.
8. Eagle RC, Jr. Eye Pathology: An Atlas and Text. 2nd ed. Philadelphia: Lippincott Williams & Willkins, 2011.
9. Xian-li S, et al. Orbital rhabdomyosarcoma: Immunoshistochemical studies of seven cases. Chin Med J 1990;103(6):485-88.
10. Furlong MA, Mentzel T, Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol 2001;14(6):595-603.
11. Raney RB, et al. The intergroup rhabdomyosarcoma study group (IRSG): Major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma 2001;5:9-15.
12 Bagdonaite L, et al. Multidisciplinary management of adult orbital rhabdomyosarcoma. Orbit 2013;32(3): 208-10.
13. Crist W, et al. The third intergroup rhabdomyosarcoma study. J Clin Oncol 1995;13(3):610-30.


