Presentation and Initial Work-up
A 76-year-old male presented with three months of persistent swelling of the right upper lid associated with burning, pain and epiphora. On questioning he complained of a mechanical superior visual field defect from his eyelid and intermittent double vision on downgaze. He denied blurry vision, flashes/floaters, pain with EOMs, headaches, pulsatile tinnitus, fevers and chills. He had been diagnosed with allergies and possible sinusitis as the etiology, and was previously treated with a course of antibiotics and a Medrol dose pack without symptomatic relief.
Medical History
His past medical history was notable for follicular lymphoma of his right arm and bronchus in 2018 (he underwent treatment with radiation and rituximab without evidence of recurrence on the most recent Positron Emission Tomography performed three months prior), prostate cancer (in remission without metastatic disease), cutaneous squamous cell carcinoma of the left brow and forehead (for which he underwent Mohs-guided resection without evidence of recurrence), hypothyroidism and hyperlipidemia. His medications included levothyroxine and atorvastatin. He had no contributory ocular history.
What is your diagnosis? What further work-up would you pursue? The diagnosis appears below.
 |
|
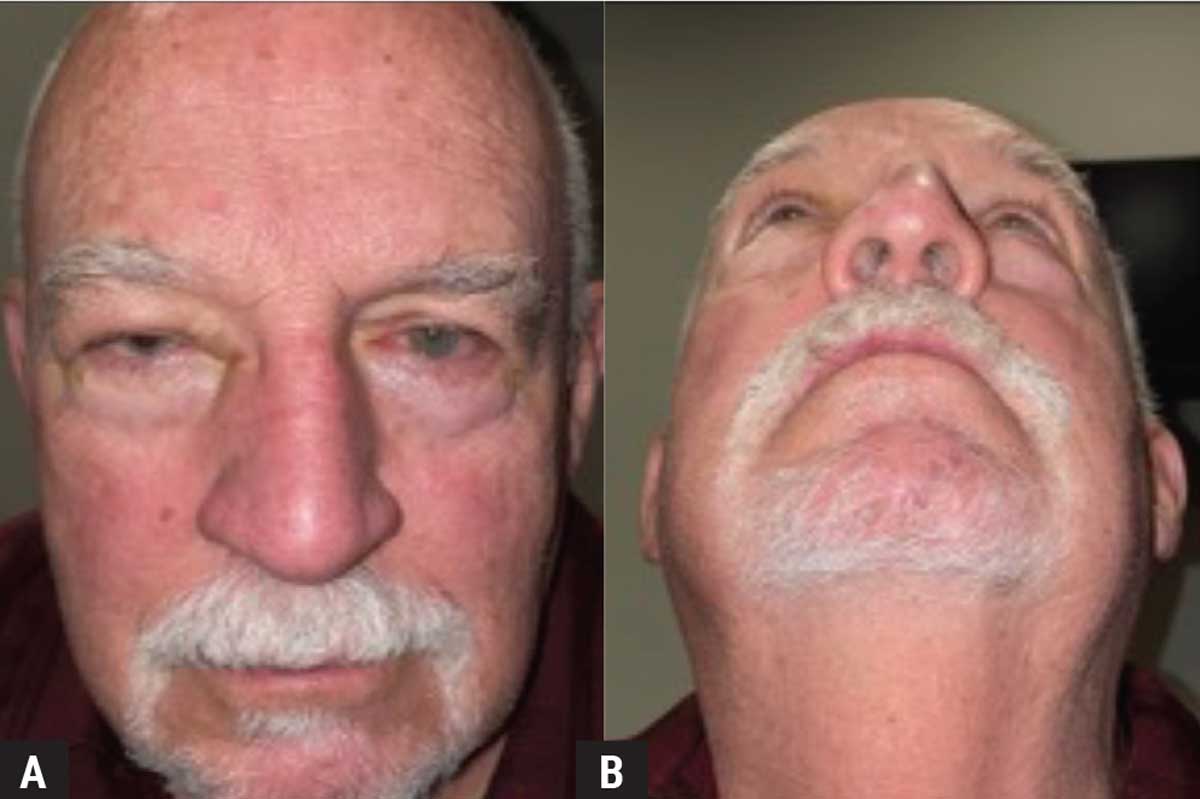
Figure 1. External photograph showing mechanical ptosis (A) and relative proptosis (B) of the right eye. |
Work-up, Diagnosis and Treatment
The patient’s periorbital swelling, mechanical ptosis, relative proptosis, chemosis and elevated intraocular pressure were concerning for an orbital process. The initial differential included inflammatory and neoplastic processes (i.e., orbital recurrence of prior lymphoma) as well as vascular etiologies such as cavernous-carotid fistula, with infectious etiologies being less likely given the duration of symptoms.
MRI of the orbits was obtained with and without gadolinium and demonstrated a 3.1 x 2.2 x 2.2 cm right intraconal mass with capsular enhancement. The mass infiltrated the muscle cone medially and encased the intraconal structures, causing local mass effect on the globe and optic nerve (Figure 2).
The patient underwent orbitotomy with diagnostic biopsy. Histopathologic evaluation demonstrated an infiltrate of neoplastic, mitotically active epithelioid cells within the skeletal muscle, arranged in sheets, strands and nests (Figure 3A, B). The neoplastic cells were positive for Melan-A, SOX10 and S100 and were negative for CD45 (pan-lymphocyte marker) and NKX3.1 (prostate carcinoma marker). This was compatible with melanoma. Pathology suggested metastatic origin given involvement of the muscle (Figure 3C).
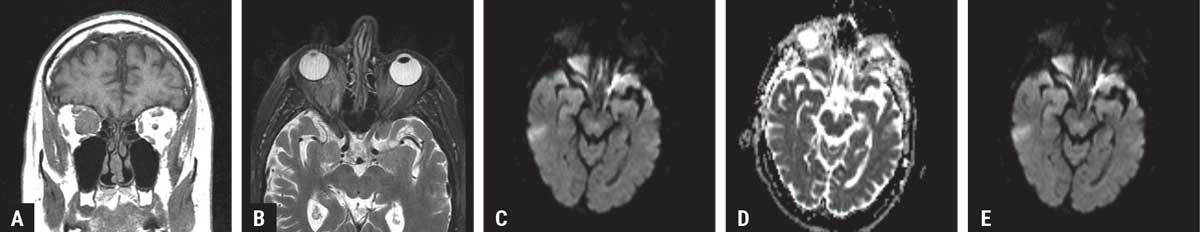 |
| Figure 2. MRI demonstrating a right intraconal mass. The signal characteristics were low in signal on T1 (A) and T2 weighted images (B). Internally, the matrix of the mass demonstrated restricted diffusion on both DWI (C) and ADC (D) maps. Following infusion of contrast material, there was homogeneous enhancement of the tumor matrix with a well-defined capsule that featured increased enhancement (E). Given the patient’s history of follicular lymphoma, orbital recurrence of lymphoma was considered as the diagnosis. |
Additional immunohistochemical studies performed for further characterization of melanoma showed that the tumor expressed nuclear PRAME and BAP1, while BRAF was negative. Genetic analysis demonstrated high mutational burden, NRAS and TERT mutations and PDL1 positivity, conferring a favorable effect from PDL1 inhibitors.
Systemic work-up was pursued with full-body imaging, including a CT-scan of the chest, abdomen and pelvis; and an MRI of the brain. All were unrevealing. A full skin exam by a dermatologist didn’t disclose any cutaneous lesions concerning for malignancy. There were no signs of a primary conjunctival, uveal or cutaneous melanocytic lesion, including oculodermal melanocytosis (nevus of Ota). Bright scan ultrasonography was consistent with the findings of the dilated fundus examination and didn’t disclose evidence of a choroidal lesion.
The patient was referred to medical oncology to discuss treatment options. Though pathology suggested metastatic origin, no primary cutaneous melanoma or other metastatic disease was found, suggesting this may be, in fact, primary orbital melanoma.
Given the paucity of data on management of primary orbital melanoma, however, there’s no consensus on the best treatment approach. The patient was offered neoadjuvant immunotherapy extrapolated from data on cutaneous and conjunctival melanoma, or exenteration with or without adjuvant radiation or immunotherapy. Given the absence of metastatic disease (confirmed on repeat PET scan) the patient elected to proceed with orbital exenteration. He underwent orbital exenteration with free flap with a multidisciplinary team of oculoplastics and head/neck surgeons. There was no evidence of cranial base invasion and negative margins were obtained at the orbital apex intraoperatively. Final pathology again showed melanoma involving the extraocular muscle and orbital soft tissue. No precursor lesion, such as blue nevus or orbital melanocytosis, was identified. The patient is currently receiving adjuvant nivolumab monthly and a surveillance PET scan every three months. On his first three-month follow-up PET scan he maintained no evidence of metastatic disease.
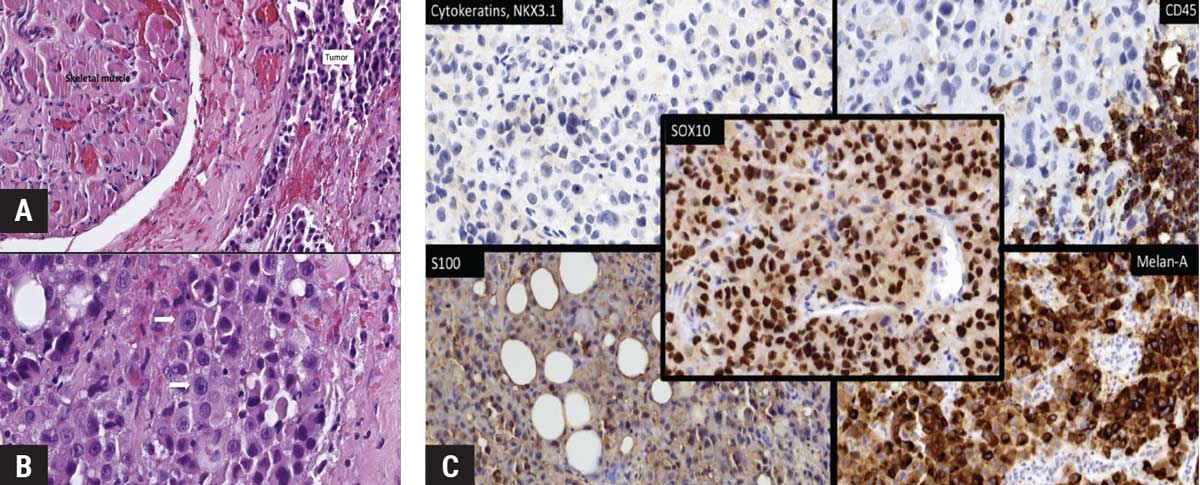 |
| Figure 3. Pathologic findings. (A) Hematoxylin-eosin stained preparation demonstrates skeletal muscle with an infiltrate of cells with epithelioid morphology arranged in sheets, strands and nests. (B) On higher magnification, the neoplastic cells have pleomorphic ovoid nuclei with prominent central nucleoli and moderate to abundant, focally densely eosinophilic cytoplasm (arrows). (C) Immunohistochemical stains show that the neoplastic cells are positive for Melan-A, S100 and SOX10. The neoplastic cells are negative for cytokeratins (CAM5.2 and AE1/A3), and pan-leukocyte marker CD45. CD45 is expressed in lymphocyte aggregates adjacent to the tumor. |
Discussion
Ocular melanoma accounts for 3 to 4 percent of all melanomas. These tumors most commonly present in the uveal tract, but can also arise in the conjunctiva and ocular adnexa.1 Orbital melanoma can occur via direct invasion from a uveal or conjunctival primary tumor or as metastasis from a distant site, usually in the setting of widespread systemic disease.1 Primary orbital melanoma (POM) is exceedingly rare.2
POM accounts for less than 1 percent of all primary orbital malignancies, with fewer than 90 cases reported in the literature to date.2-3 POM is thought to arise from melanocytes of the leptomeninges or ciliary nerves or from ectopic nests of orbital melanocytes and is often associated with the nevus of Ota (90 percent), orbital melanocytosis (50 percent) or cellular blue nevus.2-4 The most common presenting signs and symptoms are unilateral proptosis (73 percent), decreased visual acuity (32 percent), periorbital pain (14 percent), diplopia (15 percent) and palpable mass (9 percent). Imaging often shows a well-circumscribed enhancing lesion, resembling a benign tumor or AV malformation.3 On MRI, they may exhibit the unique characteristic of T1W hyperintensity and T2W hypointensity, attributed to melanin’s paramagnetic properties.5 This is not true of amelanotic melanomas, which was the case in our patient, making the biopsy diagnosis even more surprising.5
Genetic profiling of melanomas has been helpful in determining tumor origin and targeted therapy. In general, cutaneous and conjunctival melanomas harbor UV-signature mutations and exhibit a high mutational burden. Cutaneous and conjunctival melanomas are frequently enriched in MAPK, KIT, TERT, PDL-1, BRAF and/or NRAS mutations.6-7 In contrast, uveal melanoma has a lower mutational burden, with frequent mutations in GNAQ, GNA11, EIF1AX, SF3B1 and BAP1.6,8-10 Little has been reported on genetic profiling of POM. A recent study described the genetic profiles of six cases of POM, dividing them into two subgroups: uveal-melanoma-like group (expressing GNAQ, GNA11 and SF3B1 mutations) and conjunctival-melanoma-like group (expressing NRAS, TERTp).11 The authors postulated that the proximity of the tumor to the conjunctiva and thus UV damage dictates the genetic mutations.11 Newer data is demonstrating that there is more overlap between cutaneous, mucosal, conjunctival and uveal melanomas, making it difficult to establish the origin conclusively from genetic profiling alone.7,12-14
Treatment choice for POM is difficult and patients often have a poor prognosis. In a combined review of a total of 88 cases, most patients had orbital exenteration (57 percent) or excision/debulking (38 percent); half received radiation (mostly external beam or proton beam), and 14 percent received chemotherapy.3 Regardless of the selected regimen, patients often had a poor prognosis, with 36 percent developing metastasis, 15 percent developing recurrence and a 32-percent mortality at three-year follow-up.3
In the presented case, genetic profiling revealed a high mutational burden, NRAS and TERT mutations and PDL1 positivity, with no evidence of GNAQ, GNA11, SF3B1 and BAP1 mutations, compatible with a cutaneous-like or conjunctival melanoma-like mutational signature. Yet, comprehensive examination and imaging studies didn’t identify a primary site or other foci of metastatic disease. Thus, although an occurrence of primary orbital melanoma involving the EOM without pre-existing nevoid component is highly concerning for metastasis, POM emerged as a diagnosis of exclusion.
Treatment of the presented patient required a multidisciplinary approach. In addition to surgical resection, our patient also received adjuvant immunotherapy. Although CTLA-4 and PD-1 inhibitors are widely used in cutaneous melanoma, they’ve shown less promise in ocular melanomas.3 There’s a body of literature demonstrating disappointing results in uveal melanoma and mediocre results in conjunctival melanoma.15-16 It’s believed that the small mutation burden in uveal melanoma causes decreased recognition by immune cells and explains this lack of benefit to checkpoint inhibitors.1,17 However, given that the high mutational burden in our patient was similar to that of cutaneous melanoma, adjuvant immunotherapy was pursued. Experience with immune checkpoint inhibitors in POM has yet to be published.
In this review, we report a case of primary orbital melanoma presenting as unilateral lid edema and proptosis and summarize the limited literature about the clinical and histopathologic diagnosis of this rare entity. Our case offers additional insight into the use of genetic profiling for guiding diagnosis and treatment, and for understanding what prognosis to expect. Particularly, we highlight the promising role of adjuvant immunotherapy in POM.
1. Shields CL, Shields JA. Ocular melanoma: Relatively rare but requiring respect. Clin Dermatol 2009;27:1:122-33.
2. Rose AM, Luthert PJ, Jayasena CN, Verity DH, Rose GE. Primary Orbital Melanoma: Presentation, Treatment, and Long-term Outcomes for 13 Patients. Front Oncol 2017;7:316.doi: 10.3389/fonc.2017.00316.
3. Adetunji MO, McGeehan B, Lee V, Maguire MG, Briceño CA. Primary orbital melanoma: A report of a case and comprehensive review of the literature. Orbit 2020 40:6:461-469.
4. Tellado M, Specht CS, McLean IW, Grossniklaus HE, Zimmerman LE. Primary orbital melanomas. Ophthalmology 1996;103:6:929–932.
5. Uduma UF, Titalom K. Intra-orbital malignant melanoma: Role of MR imaging (a case report and literature review). Glob J Health Sci 2012;4:1:253-258.
6. Brouwer NJ, Verdijk RM, Heegaard S, et al. Conjunctival melanoma: New insights in tumour genetics and immunology, leading to new therapeutic options. Progress in Retinal and Eye Research, https://doi.org/10.1016/j.preteyeres.2021.100971. Available online May 17, 2021.
7. Gardrat S, Houy A, Brooks K, et al. Definition of biologically distinct groups of conjunctival melanomas according to etiological factors and implications for precision medicine. Cancers (Basel) 2021;13:15:3836. https://doi.org/10.3390/cancers13153836
8. Helgadottir H, Höiom V. The genetics of uveal melanoma: Current insights. Appl Clin Genet 2016;9:147-155.
9. Francis RE, Mohammed D, Jessica A, Lijka-Jones B, Sisley K. Genetics of uveal melanoma. In: Scott JF, Gerstenblith MR, eds. Noncutaneous Melanoma. Brisbane, Australia: Codon Publications, 2018:19-35.
10. Staby KM, Gravdal K, Mørk SJ, Heegaard S, Vintermyr OK, Krohn J. Prognostic impact of chromosomal aberrations and GNAQ, GNA11 and BAP1 mutations in uveal melanoma. Acta Ophthalmol 2018;96:1:31-38.
11. Mudhar HS, Doherty RE, Salvi SM, Currie ZI, Tan JH, Sisley K. Genetic profiling of primary orbital melanoma: An analysis of 6 cases with clinicopathologic correlation. Ophthalmology 2019;126:7:1045–1052.
12. Elder DE, Bastian BC, Cree IA, et al. The 2018 WHO Classification of Cutaneous, Mucosal, and Uveal Melanoma. Arch Pathol Lab Med 2020;144:4:500-522.
13. Griewank KG, Muller H, Jackett L et al. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Modern Pathology 2017;30:7:928-939.
14. Rose AM, Luo R, Radia UK, Kalirai H, Thornton S, Luthert PJ, et al. Detection of mutations in SF3B1, EIF1AX and GNAQ in primary orbital melanoma by candidate gene analysis. BMC Cancer 2018;18:1:1262.
15. Orloff, M. Clinical trials in metastatic uveal melanoma: Immunotherapy. Ocul Oncol Pathol 2021;7:3:168-176.
16. Sagiv, O, Thakar SD, Kandl TJ, et al. Immunotherapy with programmed cell death 1 inhibitors for 5 patients with conjunctival melanoma. JAMA Ophthalmol 2018;136:11:1236-1241.
17. Schank TE, Hassel JC. Immunotherapies for the treatment of uveal melanoma—history and future. Cancers (Basel). 2019;11:8:1048.


