The heterogeneous group of inflammatory chorioretinopathies that make up the disease entity commonly known as “white dot syndrome” can result in multiple visual and anatomic sequelae, and can be challenging to diagnose due to their rarity and varying pathogenesis and other factors. This review will detail each disease separately and cover predisposing conditions and pathogenesis, genetic factors, clinical features and treatments.
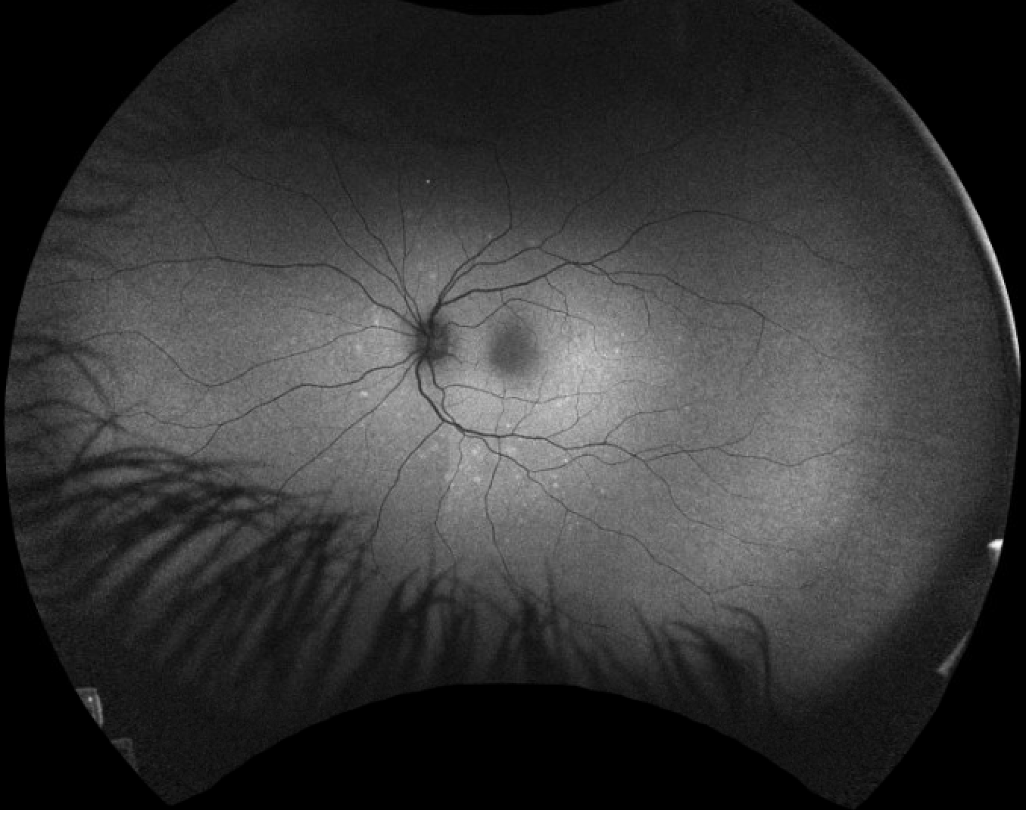 |
| Figure 1. A 19-year-old female presented with photopsias and blurred vision. Fundus examination revealed a slight granularity to the macula, composed of pinpoint, yellow-white lesions. These are demonstrated on autoflourescence with a hyperfluorescent pattern. |
WDS Background
The white dot syndromes group includes a heterogeneous mix of inflammatory chorioretinopathies. Patients typically present with blurred vision, scotomas, photopsias and floaters. On examination, the main commonality between these disorders lies in the presence of white, yellow-white, or gray-white lesions involving the outer retina, retinal pigment epithelium and choroid. The white dot syndrome category includes: multiple evanescent white dot syndrome (MEWDS); acute posterior multifocal placoid pigment epitheliopathy (APMPPE); idiopathic multifocal choroiditis (IMFC); punctate inner chorioretinopathy (PIC); serpiginous chorioretinopathy; acute zonal occult outer retinopathy (AZOOR); and birdshot chorioretinopathy (BSCR).1
These diseases are relatively rare, with an estimated incidence of 0.45 in 100,000 per year.2 The rarity of the condition makes it difficult to conduct studies assessing the predisposing factors for disease development; thus, the exact pathogenesis of these syndromes is undetermined. Patients often describe a viral prodrome, but the WDS have also been linked to a variety of other inciting factors, including—but not limited to—bacterial infections, vaccinations and certain haplotypes.3
Initial reports believed that all white dot syndromes fell on a common inflammatory spectrum and shared a similar pathogenesis.4 Recently, multi-modal imaging has improved our understanding of the pathophysiology of these diseases, and it’s become evident that not all of these conditions may fit along the same continuum. In particular, MEWDS, APMPPE, IMFC, PIC and serpiginous may fit into one group, while AZOOR and BSCR have distinct inflammatory profiles.1
Multiple Evanescent White Dot Syndrome
MEWDS has been linked to multiple exposures, the two most common of which include viruses and vaccinations, such as the Epstein-Barr, hepatitis A and B, human papilloma and herpesviridae family.5,6 One of the proposed mechanisms of MEWDS explains the phenomenon as an immune reaction to a viral infection, or the result of a complex interplay of genetic and environmental factors. Three main hypotheses exist to explain the pathophysiology of MEWDS based on in-depth imaging analysis: 1) hypoperfusion of the choriocapillaris leading to ischemic damage to overlying tissue; 2) immune damage of the RPE causing photoreceptor decline; or an immune attack targeting photoreceptors themselves.
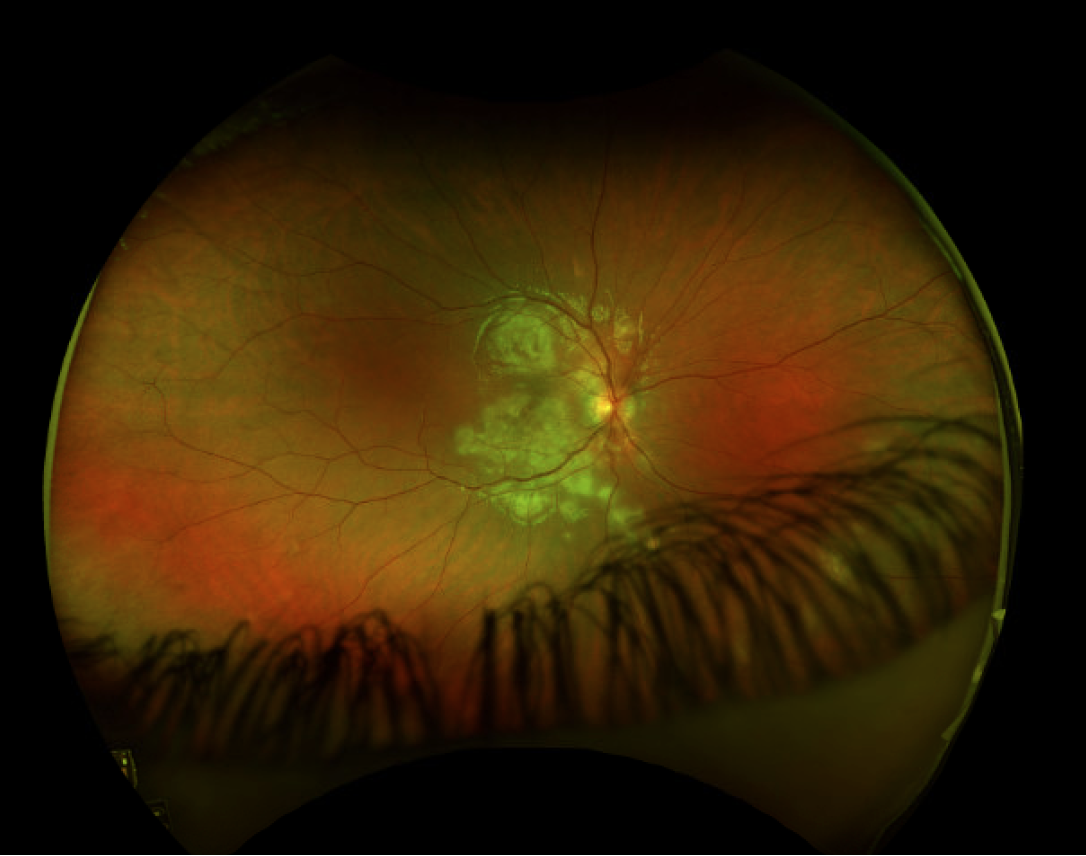 |
| Figure 2. A 10-year-old male presents with blurred vision and creamy, yellow-white lesions throughout the fundus consistent with APMPPE. Given the widespread distribution, the patient was started on oral steroids, with resolution of the lesions. |
MEWDs has traditionally been classified as monophasic and unilateral, but recent reports have shown evidence of it being an asymmetrical, bilateral process, with a recurrence rate of about 10 percent.7-9 There can be no overlying inflammation but, occasionally, mild intraocular inflammation exists, with typical findings including vitritis or optic nerve edema.10 An enlargement of the blind spot may also occur. Interestingly, many initial cases of MEWDS were wrongly categorized as acute idiopathic blind spot enlargement (AIBSE).11 On dilated examination, small, discrete or confluent yellow-white dots are visible, usually ranging in size from 100 μm to 200 μm through the outer retina or the RPE. These are typically located in the macula but they can also involve the mid-periphery.12 Additionally, a macular granular change can occur; in rare cases this granularity may be the only finding if the deep retinal or RPE dots have resolved.
Likely with all white dot syndromes, multi-modal imaging is instrumental in making the diagnosis. Optical coherence tomography often reveals photoreceptor damage, and fluorescein angiography shows retinal pigment epithelium abnormalities. These most often present with a typical wreath pattern of early hyperfluorescence and late staining of the lesions, while indocyanine green angiography is helpful, as it reveals more hypocyanescence lesions than appreciated on examination. Autofluorescence can show areas of hyperfluorescence that correlate to the lesions seen on ICGA, and the presence of hyperfluorescence can indicate current or recent activity (Figure 1). The enlarged blind spot can be confirmed with visual field testing.
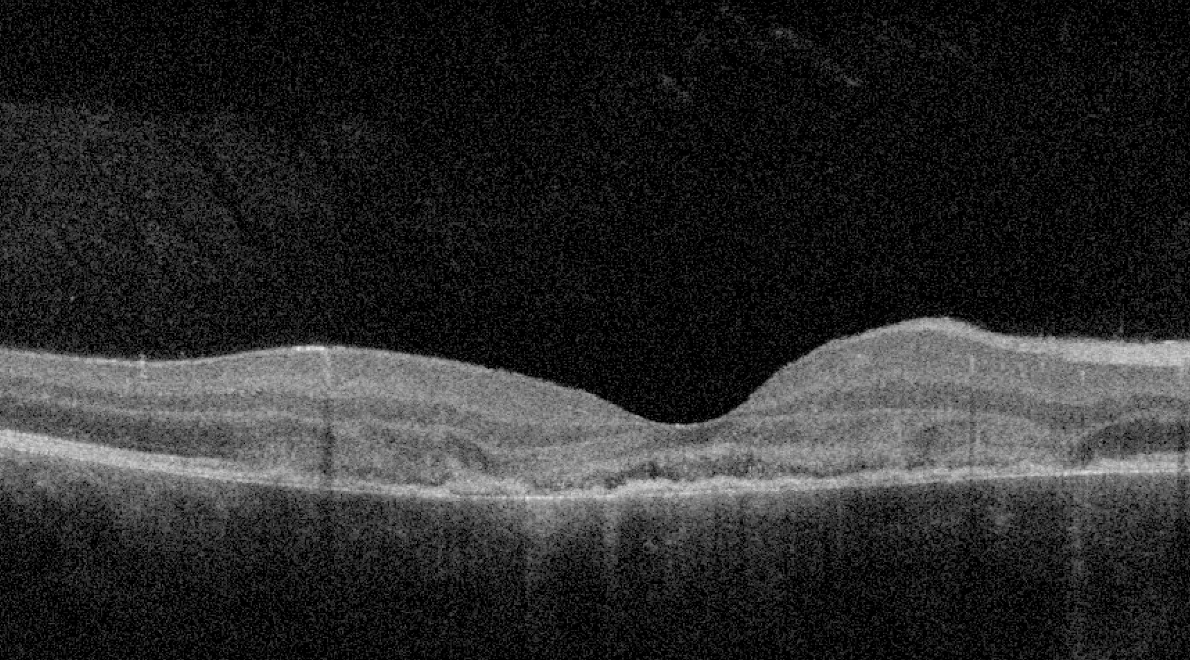 |
| Figure 3. OCT shows outer retinal disorganization and photoreceptor loss, consistent with changes from APMPPE. |
Acute Posterior Multifocal Placoid Pigment Epitheliopathy
Similarly to MEWDS, APMPPE has been connected to infectious primers, including viruses, tuberculosis or vaccinations.13 Other immune conditions linked to APMPPE include psoriasis, sarcoidosis, erythema nodosum, granulomatosis with polyangitis, polyarteritis nodosa and diabetes.14 The most recent literature identifies the choriocapillaris as the primary site of immune attack, which results in destruction of the overlying photoreceptors.14
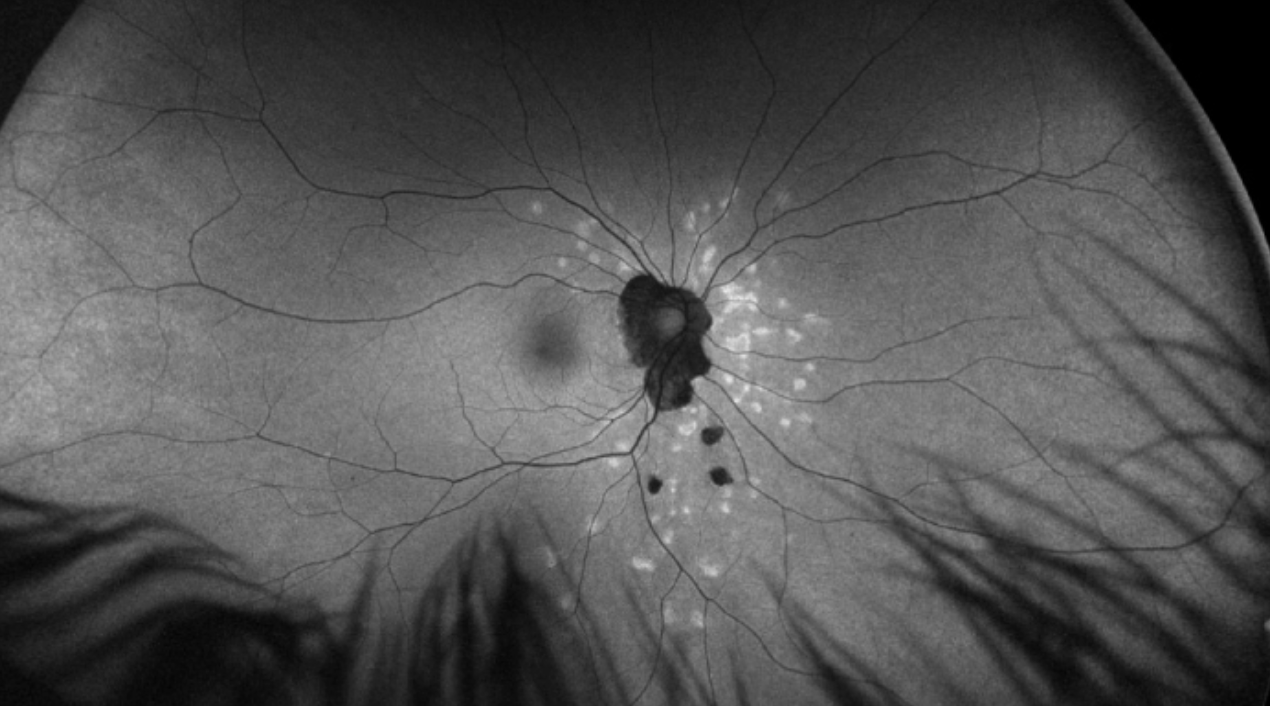 |
| Figure 4. Multiple chorioretinal lesions throughout the fundus in a 27-year-old female consistent with idiopathic multifocal choroiditis. Hyperfluorescent areas demonstrate areas of recent activity. |
APMPPE often presents as an acute, bilateral disorder in young healthy adults. Fundoscopy shows multifocal, creamy to yellowish placoid lesions in the posterior pole10 (Figure 2). OCT confirms disturbance of the outer retina and photoreceptors, along with RPE hyperreflectivity (Figure 3). Early phases of fluorescein angiography reveal hypofluorescent lesions, which later become hyperfluorescent, and ICGA demonstrates hypocyanescence in both early and late phases.15 Cases are usually self-limited and don’t require therapy, although the use of oral steroids have been advocated, especially in foveal threatening disease. Despite being traditionally characterized as a self-resolving disease, there are variations of APMPPE that have a worse prognosis, including persistent placoid maculopathy and relentless placoid chorioretinitis. Their clinical course is prolonged and characterized by multiple relapses.16
Of note, patients who present with signs of APMPPE should also be assessed for neurological symptoms. Clinicians should have a low threshold for obtaining neurological imaging. In one review, half of patients with neurological symptoms in the setting had cerebral vasculitis. Other, more unusual complications include meningioencephalitis, viral meningitis, cavernous sinus thrombosis, and sixth-nerve palsy.17 Rarely, death can result from cerebrovascular complications.18
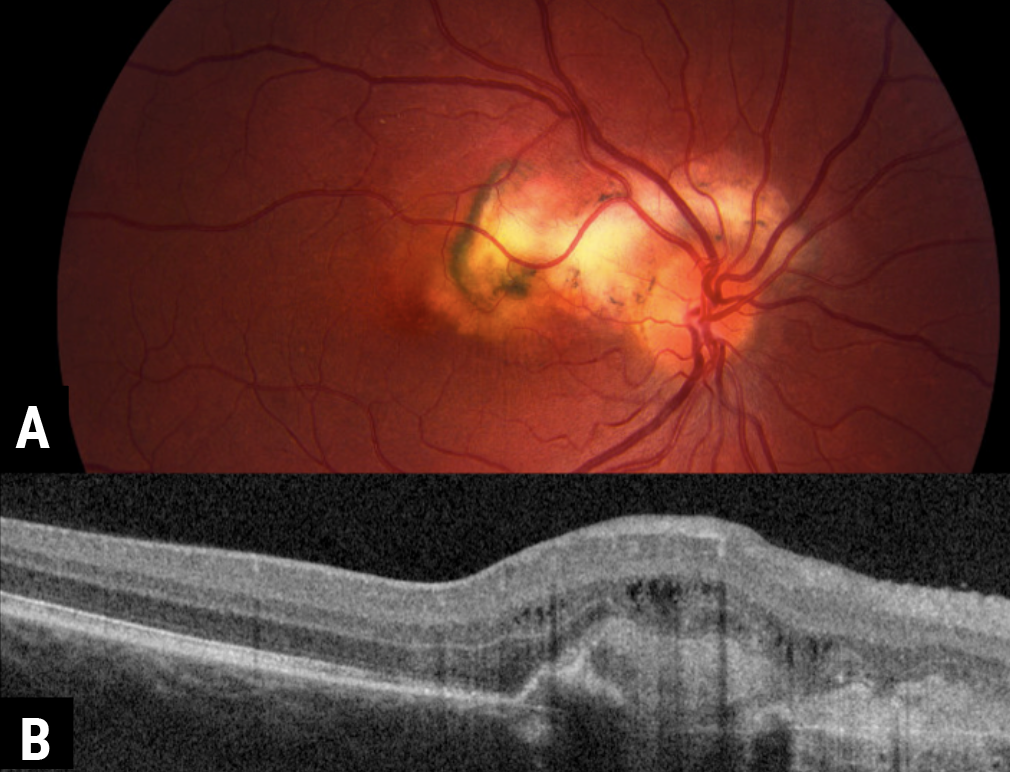 |
| Figure 5. A) A 34-year-old female presents with blurred vision, with fundus examination showing peripapillary scarring extending into the macula consistent with serpiginous choroidopathy. B) On OCT, a choroidal neovascular membrane formed at the peripheral edge, requiring anti-VEGF injections. |
IMFC and PIC
Both idiopathic multifocal choroiditis and punctate inner chorioretinopathy have been linked to a viral prodrome or vaccination, but a definitive cause remains elusive. Characteristically, IMFC is chronic and recurrent, with unilateral or bilateral involvement. The hallmark of this disease requires subretinal lesions located throughout the macula and peripheral retina, with possible overlying vitreous inflammation. These lesions range from 50 to 350 μm and are yellow to yellow-white, while hyperfluorescence can demonstrate areas of activity19 (Figure 4). Vision loss occurs when these lesions result in chorioretinal atrophy or choroidal neovascular membrane (CNVM) development.10 The OCT demonstrates loss of photoreceptors, with retinal architectural changes tracking to the inner retina. Optical coherence tomography angiography can also be very helpful in these cases, and confirm dropout of mid-sized vessels in the choriocapillaris. Additionally, in patients presenting with bilateral MEWDS, or MEWDS that recurs with chorioretinal scarring, IMFC should be considered, as it can initially present as MEWDS. 12
PIC shares many of the same features of IMFC, and differentiation of the two may be difficult. In classic cases, traditional PIC lesions are located in the posterior pole and range from 100 to 300 μm. The disease is almost always bilateral, even if the patient initially presented with unilateral disease.20 In contrast to IMFC, PIC has no inflammation, but does carry with it a high risk of CNVM development.
Serpiginous Chorioretinopathy
Much like the other white dot syndromes, a viral prodrome has been described before symptom onset with serpiginous chorioretinopathy. PCR testing of the aqueous humor in these eyes has revealed herpes virus in a subset of patients, but this hasn’t been universally true.21 Serpiginous chorioretinitis most often has a geographic yellow to yellow-grey choroiditis that extends from the optic nerve with no to very little overlying inflammation (Figure 5A). Recurrences usually occur at the margins of prior scarring, and concurrent active and inactive lesions typify the disease. Reactivation is common (50 percent within five years), and macular involvement occurs in approximately 90 percent of untreated patients.22 In cases where the serpiginous disease isn’t treated, the chorioretinitis typically burns out over two decades, but leaves extensive scarring and fibrosis behind.22 Optical coherence tomography demonstrates outer retinal disorganization and hyperreflectivity of the RPE, and hyperfluorescent areas can delineate areas of early disease activity or recurrence.23 CNV can also develop at the margin of impacted tissue (Figure 5B).
While the above five diseases (MEWDS, APMPPE, IMFC, PIC, serpiginous) all involve occlusion or non-perfusion to the choroid, differences exist in the caliber of vessels involved. MEWDS is thought to preferentially impact the end capillary vessels, while APMPPE, IMFC and PIC involve the mid-sized choriocapillaris vessels, and serpiginous is hypothesized to destroy the larger choriocapillaris.24 Of note, certain researchers believe that IMFC and PIC are variations of the same disease, and that MEWDS may represent early IMFC if it recurs.25
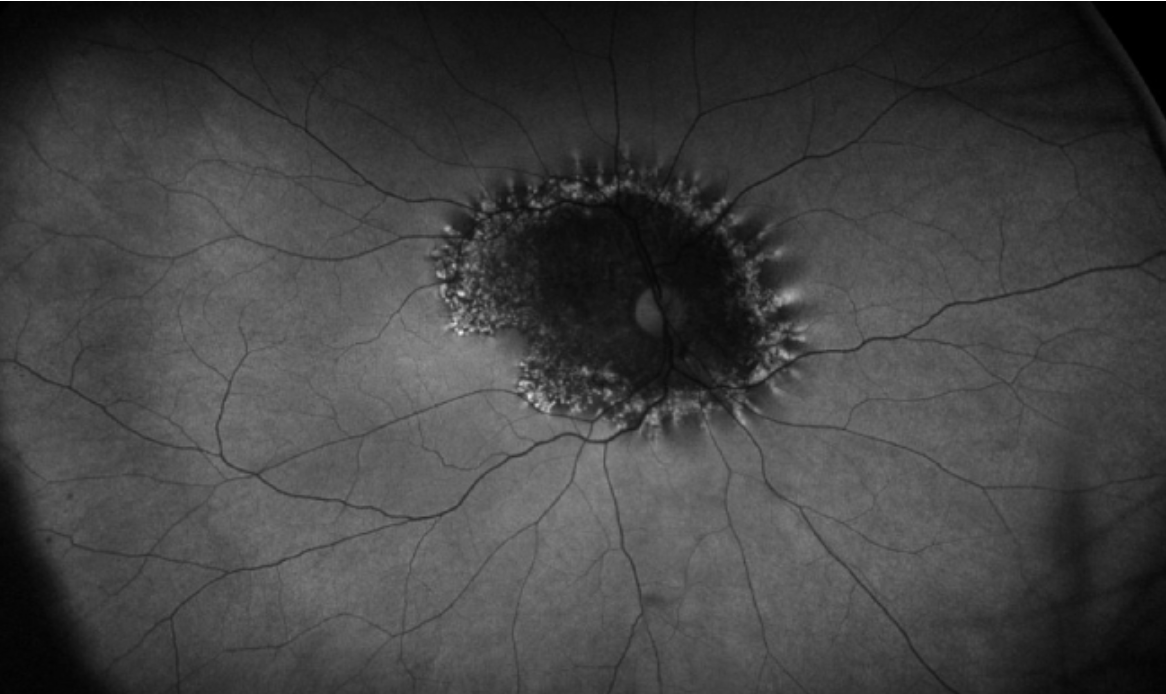 |
| Figure 6. A 47-year-old male presents with bilateral blurred vision, and fundus examination shows a faintly pigmentary changes around the nerve. Autofluorescence shows the typical trizonal pattern, including inner hypofluorescence, a rim of hyperfluorescence, and normal retina outside of the area of involvement. |
Acute Zonal Occult Outer Retinopathy (AZOOR)
Unlike the previously described diseases that mainly involve the choriocapillaris, AZOOR refers to an inflammation aimed at the photoreceptors, but the exact mechanism is poorly understood. Some believe that a viral infection also predisposes the body to attack the photoreceptors but this hasn’t been proven.26 The disease mainly impacts young to middle aged myopic women, with 75 percent developing bilateral symptoms. Initially, the fundus examination may be inconspicuous, but ultimately a faint, white to greyish white line delineating the normal area from the involved retina may appear.27 It’s rare to have overlying inflammation in the vitreous. The OCT shows photoreceptor destruction, and fluorescein angiography either demonstrates leakage or staining over these areas. Fundus autofluorescence can reveal a stereotypical trizonal pattern: hypofluorescence over the area that has already been involved, a hyperfluorescence area at the border, and normal pattern after the area of AZOOR (Figure 6). AZOOR usually stabilizes at six months, and patients often maintain good vision. You can consider treatment with local or oral steroids however, especially in foveal threatening disease or recurrence. In a large report, the recurrence rate was documented in 15 percent of patients.28
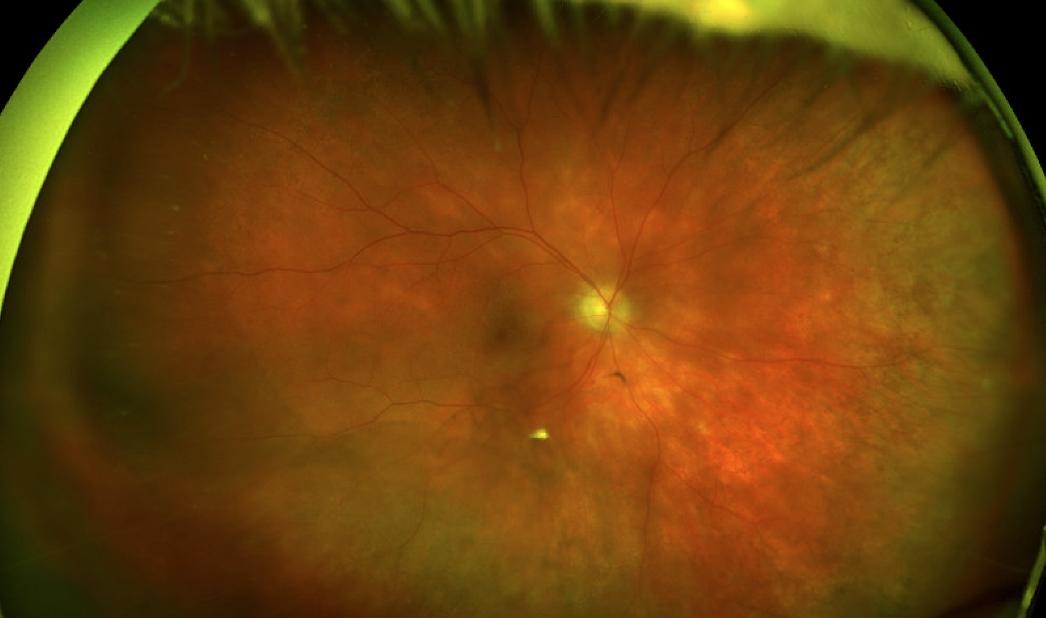 |
| Figure 7. A 63-year-old woman presents with a two-year history of blurred vision. Fundus examination revealed 1+ vitritis and creamy yellow-white lesions throughout the fundus, concentrated more inferotemporally. |
Birdshot Chorioretinopathy (BSCR)
BSCR is hypothesized to be an inflammation of both the retina and choroid and linked to the haplotype HLA-A29. This link is so strong that, in a patient with negative HLA-A29, testing should raise the possibility of an alternative diagnosis. It’s also important to note that HLA-A29 positivity is already present in 7 percent of the general population; therefore, its presence does guarantee a diagnosis of birdshot. Recent studies have also shown a link between BSCR and endoplasmic reticulum aminopeptidase 2 (ERAP2). ERAP2, like HLA-A29, is involved in the antigen presentation process, and the increase of this may activate the immune response in those with the HLA–A29 phenotype, but the true mechanism remains poorly understood.29
BSCR presents with the typical range of WDS symptoms, in addition to visual field constriction. Unlike most of the other WDS, patients tend to develop BSCR later in life. Fundus examination reveals vitritis overlying scattered choroidal creamy yellow-white lesions, with a high concentration located inferonasally (Figure 7). Fluorescein angiography can reveal disc edema, macular leakage or retinal vascular leakage. Like other WDS, ICGA can reveal choroidal lesions that aren’t seen on fundus photography. OCT can show macular edema, photoreceptor and ellipsoid zone disruption as well as choroidal thinning. Visual field monitoring is important, since the earliest signs can be peripheral visual loss. Given its chronic and progressive nature, treatment is important in patients with BSCR. The treatment regimen is usually stepwise, initially beginning with local or oral steroids, with subsequent progression to steroid-sparing therapy.30
In conclusion, prompt recognition of these diseases is essential to obtain the best visual outcomes. Multi-modal imaging has provided new insights into the classification and understanding of the pathophysiology of these diseases. In certain cases, the patient can be watched, but in severe cases, involving foveal-threatening disease, or complications such as choroidal neovascular membranes, treatment is necessary.
Dr. Arepalli is an assistant professor in the Vitreoretinal and Uveitis Service at Emory University.
Dr. Regillo is the director of the Retina Service of Wills Eye Hospital, a professor of ophthalmology at Thomas Jefferson University School of Medicine and the principle investigator for numerous major international clinical trials.
Dr. Yonekawa is an assistant professor of ophthalmology at Sidney Kimmel Medical College at Thomas Jefferson University. He serves on the Education Committee of the American Society of Retina Specialists and on the Executive Committee for the Vit Buckle Society, where he is also the vice president for academic programming.
1. Mount GR, Kaufman EJ. White dot syndromes. StatPearls. Treasure Island (FL): StatPearls Publishing 2022.
2. Abu-Yaghi NE, Hartono SP, Hodge DO, Pulido JS, Bakri SJ. White dot syndromes: A 20-year study of incidence, clinical features, and outcomes. Ocular immunology and inflammation 2011;19:6:426-430.
3. Crawford CM, Igboeli O. A review of the inflammatory chorioretinopathies: The white dot syndromes. ISRN inflammation 2013;2013:783190.
4. Ben Ezra D, Forrester JV. Fundal white dots: The spectrum of a similar pathological process. The British journal of ophthalmology 1995;79:9:856-860.
5. Yang CS, Hsieh MH, Su HI, Kuo YS. Multiple evanescent white dot syndrome following acute Epstein-Barr virus infection. Ocular immunology and inflammation 2019;27:2:244-250.
6. Haw YL, Yu TC, Yang CS. A CARE-compliant article: A case report of possible association between recurrence of multiple evanescent white dot syndrome and the Herpesviridae family. Medicine 2020;99:15:e19794.
7. Veronese C, Maiolo C, Morara M, Armstrong GW, Ciardella AP. Bilateral multiple evanescent white dot syndrome. International Ophthalmology 2018;38:5:2153-2158.
8. Bosello F, Westcott M, Casalino G, et al. Multiple evanescent white dot syndrome: Clinical course and factors influencing visual acuity recovery. British Journal of Ophthalmology 2022;106:1:121-127.
9. Ramakrishnan MS, Patel AP, Melles R, Vora RA. Multiple evanescent white dot syndrome: Findings from a large Northern California cohort. Ophthalmology Retina 2021;5:9:850-854.
10. Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of Ophthalmic Inflammation and Infection 2021;11:1:32.
11. Fletcher WA, Imes RK, Goodman D, Hoyt WF. Acute idiopathic blind spot enlargement. A big blind spot syndrome without optic disc edema. Archives of Ophthalmology 1988;106:1:44-49.
12. Papasavvas I, Mantovani A, Tugal-Tutkun I, Herbort CP, Jr. Multiple evanescent white dot syndrome (MEWDS): Update on practical appraisal, diagnosis and clinicopathology; A review and an alternative comprehensive perspective. Journal of Ophthalmic Inflammation and Infection 2021;11:1:45.
13. Gonome T, Suzuki Y, Metoki T, Takahashi S, Nakazawa M. Acute posterior multifocal placoid pigment epitheliopathy and granulomatous uveitis following influenza vaccination. American Journal Of Ophthalmology Case Reports 2016;4:60-63.
14. Testi I, Vermeirsch S, Pavesio C. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE). Journal of Ophthalmic Inflammation and Infection 2021;11:1:31.
15. Mrejen S, Sarraf D, Chexal S, Wald K, Freund KB. Choroidal involvement in acute posterior multifocal placoid pigment epitheliopathy. Ophthalmic Surgery, Lasers & Imaging Retina 2016;47:1:20-26.
16. Mirza RG, Jampol LM. Relentless placoid chorioretinitis. International ophthalmology clinics 2012;52:4:237-242.
17. Algahtani H, Alkhotani A, Shirah B. Neurological manifestations of acute posterior multifocal placoid pigment epitheliopathy. Journal of Clinical Neurology (Seoul, Korea) 2016;12:4:460-467.
18. El Sanhouri A, Sisk RA, Petersen MR. Mortality from cerebral vasculitis associated with rapid steroid taper during treatment of acute posterior multifocal placoid pigment epitheliopathy. Archives of Ophthalmology 2012;130:7:935-937.
19. Papasavvas I, Neri P, Mantovani A, Herbort CP, Jr. Idiopathic multifocal choroiditis (MFC): Aggressive and prolonged therapy with multiple immunosuppressive agents is needed to halt the progression of active disease. An offbeat review and a case series. Journal of Ophthalmic Inflammation and Infection 2022;12:1:2.
20. Gerstenblith AT, Thorne JE, Sobrin L, et al. Punctate inner choroidopathy: A survey analysis of 77 persons. Ophthalmology 2007;114:6:1201-1204.
21. Rodman J, Pizzimenti J. Serpiginous choroiditis in a herpes-positive patient. Optometry and Vision Science 2011;88:6:776-780.
22. Weiss H, Annesley WH, Jr., Shields JA, Tomer T, Christopherson K. The clinical course of serpiginous choroidopathy. Am J Ophthalmol 1979;87:2:133-142.
23. Nazari Khanamiri H, Rao NA. Serpiginous choroiditis and infectious multifocal serpiginoid choroiditis. Survey of Ophthalmology 2013;58:3:203-232.
24. Papasavvas I, Mantovani A, Herbort CP. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE): A comprehensive approach and case series: Systemic corticosteroid therapy is necessary in a large proportion of cases. Medicina (Kaunas, Lithuania) 2022;58:8.
25. Lages V, Mantovani A, Papadia M, Herbort CP. MEWDS is a true primary choriocapillaritis and basic mechanisms do not seem to differ from other choriocapillaritis entities. Journal of Current Ophthalmology 2018;30:4:281-286.
26. Herbort CP, Arapi I, Papasavvas I, Mantovani A, Jeannin B. Acute zonal occult outer retinopathy (AZOOR) results from a clinicopathological mechanism different from choriocapillaritis diseases: A multimodal imaging analysis. Diagnostics (Basel, Switzerland) 2021;11:7.
27. Tsang SH, Sharma T. Acute zonal occult outer retinopathy (AZOOR) and related diseases. Advances in Experimental Medicine and Biology 2018;1085:233-237.
28. Monson DM, Smith JR. Acute zonal occult outer retinopathy. Survey of Ophthalmology 2011;56:1:23-35.
29. Donvito B, Monnet D, Tabary T, et al. A New HLA extended haplotype containing the A*2910 allele in birdshot retinochoroidopathy: Susceptibility narrowed to the HLA molecule itself. Investigative Ophthalmology & Visual Science 2010;51:5:2525-2528.
30. Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Birdshot Chorioretinitis. Am J Ophthalmol. 2021;228:65-71.