
Presentation
A 45-year-old Asian female presented with a chief complaint of right upper lid swelling for two years. She stated that it had been getting worse over the last six months, and she could now palpate a "marble-sized cyst" in her right upper eyelid. She denied pain, redness or any vision fluctuation. She noted that she had developed intolerance to wearing her contact lenses in her right eye.
Medical History
She denied any past ocular history. Her past medical history was significant for renal failure secondary to a hypertensive crisis and was status post kidney transplant. She was taking cyclosporine 300 mg daily and prednisone 10 mg every other day, as well as tenormin and hydralazine. Her family history was noncontributory and social history unremarkable. Review of systems was otherwise negative.
Examination
On examination, her best corrected visual acuity was 20/30 OD and 20/20 OS. Her pupils were round, equal, briskly reactive and without a relative afferent pupillary defect. Extraocular motility and confrontational visual fields were full in both eyes. Intraocular pressures were 12 mmHg OU. There was minimal proptosis of 1 to 2 mm on the right.
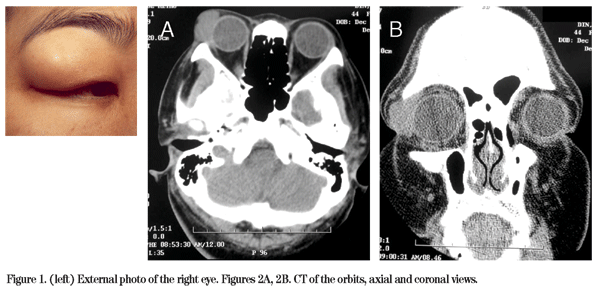
External examination of the right eye revealed a palpable mass in the superotemporal orbit (See Figure 1). Slit-lamp biomicroscopy revealed superficial punctate keratopathy OD and was normal OS. Computed tomography of the orbit was obtained and disclosed a 2 x 1.5 cm soft tissue density mass in the area of her right lacrimal gland (See Figures 2A and 2B).
Diagnosis, Workup and Treatment
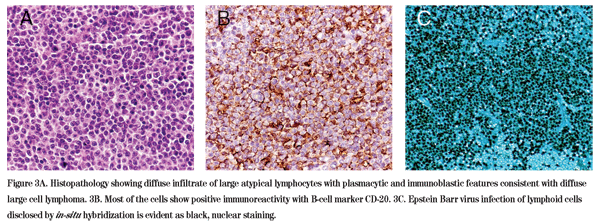
The differential diagnosis for a slowly progressive, painless unilateral lacrimal gland mass includes epithelial tumors of the lacrimal gland including pleomorphic adenoma and adenoid cystic carcinoma, lymphoma, fibrous histiocytoma, metastatic tumor and sarcoidosis. Given the patient's history of renal transplantation and immunosuppression, post-transplant lymphoproliferative disorder specifically was also in the differential. The decision was made to perform an excisional biopsy of the right lacrimal gland mass. The biopsy revealed an intense diffuse infiltrate of atypical CD-20 positive B lymphocytes with plasmacytic and immunoblastic features consistent with a diffuse large B-cell non-Hodgkin's lymphoma (See Figure 3A). Seventy-five percent of the lymphocytes showed positive intranuclear in-situ hybridization for Epstein-Barr virus, consistent with the diagnosis of post-transplant lymphoproliferative disorder (See Figure 3B and 3C).
A systemic workup that included a CT of the chest, abdomen and pelvis was negative. The patient's immunosuppressive medications were reduced since she was considered to be at low risk for rejection, as the organ donor was her sister and the transplantation had been performed many years previously. There has been no evidence of recurrence on serial imaging following mass excision and reduction of immunosuppression.
Discussion
Post-transplant lymphoproliferative disorder is a complication of solid-organ or bone-marrow transplantation. The term PTLD was initially used to describe the proliferation of B-lymphocytes resulting in a spectrum of disorders in immunosuppressed patients. It is usually caused by Epstein-Barr virus (EBV) infection of B-cells with subsequent proliferation. Although T-cell proliferations after transplantation have been described as well, they are not related to EBV infection and occur much less frequently. Cardiac transplantation is associated with the highest rate of PTLD, with a reported incidence of 4.9 to 13 percent , followed by liver transplantation (2 percent), and renal transplantation (1.9 percent in one study). The incidence of PTLD after renal-transplantation is higher in children (10.1 percent) than adults (1.2 percent). It is presumed the higher incidence of PTLD after cardiac transplantation is related to the higher levels of immunosuppression required to prevent rejection. It has been found that reducing immunosuppressive therapy in such patients ameliorates the lymphoproliferative lesions, but can also result in the rejection of the graft.
The clinical presentation of PTLD most typically includes fever, lymphadenopathy, gastro-intestinal symptoms and infectious mononucleosis-like symptoms, although our patient did not complain of any of these symptoms. The organs affected most frequently by PTLD are lymph nodes, liver, lung, kidney, bone marrow, small intestine, spleen, CNS and large intestine, in decreasing order of frequency. Ocular involvement is very rare, although there have been case reports of PTLD presenting as granulomatous uveitis with iris nodules in a 9-year-old, a chorioretinal mass in a 59-year-old, and bilateral gelatinous conjunctival lesions in an 8-year-old.
The lesions of PTLD form a pathologic spectrum of lymphoproliferative disorders. Early lesions show reactive plasmacytic hyperplasia and often present like infectious mononucleosis. The spectrum also includes polyclonal polymorphic B-cell hyperplasia which also depends on continued replication of EBV. In some cases a cytogenetic event or selection confers malignant growth potential on an EBV-infected B cell, leading to the outgrowth of a clone of malignant cells. This malignant transformation results in monomorphic monoclonal PTLD, including T-cell neoplasms (rare) and B-cell neoplasms, such as diffuse large B-cell lymphoma, Burkitt lymphoma, plasma cell myeloma, and plasmacytoma-like lesions.
There are two types of PTLD. The first occurs less than one year after transplantation, is more often polyclonal in cell morphology, and is of acute-onset, with variable prognosis. The second occurs more than one year post-transplantation, is usually monoclonal and indolent, and is more circumscribed in presentation, without systemic complaints. Lesions that arise more than five years after transplantation have a lower occurrence of EBV infection. The type of immunosuppression and the total immunosuppressive burden also appear to affect the incidence of PTLD and the time to onset, with more immunosuppression correlating with an increased incidence of PTLD. Other factors associated with poor prognosis include T-cell origin, monoclonality, multiple sites of involvement, non-detection of EBV and mononucleosis-like presentation.
Treatment for PTLD is decreasing the level of immunosuppression, although a fine balance is required to avoid graft rejection. This is easier to do in renal PTLD compared to cardiac PTLD where graft rejection could mean imminent death. Therapy also tends to be more successful in the first year post-transplantation. For focal lesions, in addition to reduced immunosuppression, local resection or radiation therapy are options that are possibly curative. The use of acyclovir and ganciclovir has been studied but has proven ineffective. The use of chemotherapy is considered to be a last resort since it enhances the immunosuppression. In our patient, a combination of local resection and reduced immunosuppression was curative.
Dr. Gold would like to thank Mary Stefanyszyn, MD, and Jacqueline Carrasco, MD, of the Oculoplastics Service, and Ralph Eagle, MD, Ocular Oncology Service, at Wills Eye Institute for their assistance with this case.
Gottschalk S, Rooney CM, Heslop HE. Post-transplant lymphoproliferative disorders. Annu Rev Med 2005;56:29-44.
Hauke R, Smir B, Greiner T, Bierman P, et al. Clinical and pathological features of posttransplant lymphoproliferative disorders: Influence on survival and response to treatment. Ann Oncol 2001;12:831-4.
Leblond V, Dhedin N, Mamzer Bruneel MF, Choquet S, et al.. Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. J Clin Oncol 2001;19:772-8.
Nalesnik MA, Makowka L, Starzl TE. The diagnosis and treatment of posttransplant lymphoproliferative disorders. Curr Probl Surg 1988;25:367-472.
Starzl TE, Nalesnik MA, Porter KA, Ho M, et al. Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. Lancet 1984;1:583-7.
Swinnen LJ, Costanzo-Nordin MR, Fisher SG, O'Sullivan EJ, et al. Increased incidence of lymphoproliferative disorder after immunosuppression with the monoclonal antibody OKT3 in cardiac-transplant recipients. N Engl J Med 1990;323:1723-8.


