 |
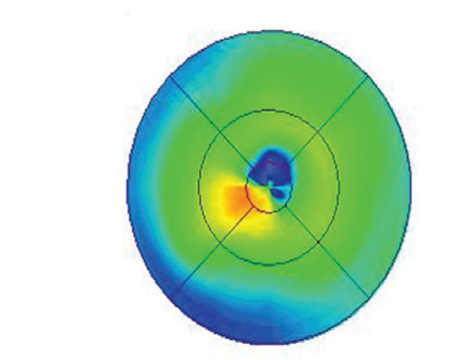
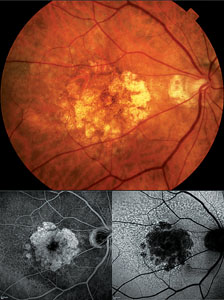
| Figure 1. Color fundus photograph (top) with corresponding fluorescein angiogram (bottom left) and autofluorescence (bottom right) images of a patient with geographic atrophy. |
Over the past several years, however, a significant surge has occurred in research focused on treatments for dry AMD. Treatment strategies have primarily centered on three major pathways: first, reduction of oxidative stress by antioxidant therapy; second, prevention of photoreceptor and RPE loss, which includes neuroprotection, reduction of toxic by-product accumulation, and modulation of the visual cycle; and third, suppression of inflammation.2 This review will describe the various drugs and therapies for dry AMD under investigation employing these specific treatment strategies.
Antioxidant Therapy
Ample evidence has established oxidative stress as an important contributor in the pathogenesis of AMD.1-4 The formation of reactive oxygen species not only appears to damage the retinal pigment epithelium but also impairs local complement inhibition, exacerbating local inflammatory processes that contribute to progression of AMD. Additionally, a polymorphism in mitochondrial DNA (A4917G), which is involved in both DNA repair and control of oxidative stress, has been found to increase the risk of AMD.5 In 2001, the Age-Related Eye Disease Study was the first major randomized prospective study that showed daily high doses of the antioxidants beta-carotene, vitamin C and E, zinc and copper decreased the risk of progression to advanced AMD in patients with intermediate forms of AMD. In 2006, the National Eye Institute began a follow-up study, the AREDS2 study, comparing the additional supplements of lutein and zeaxanthin, as well as the omega-3 fatty acids, combined with or separate from the original AREDS formula. Investigators are also examining the effects of decreasing the dosage of beta-carotene and zinc in the original AREDS formula. Recruitment has been completed and early results should be announced soon.
One of the most interesting treatment strategies for dry AMD is modulation of the visual cycle. By disrupting the conversion of retinol to rhodopsin, the key metabolite of phototransduction, toxic waste products such as lipofuscin and A2E are decreased in the RPE. Treatments along this pathway include ACU-4429 and fenretinide. ACU-4429 is a small, nonretinoid molecule that targets RPE65, one of the enzymes involved in converting all-trans retinol to all-cis retinal within the RPE. A Phase I trial was completed and showed oral ACU-4429 to be safe and well-tolerated in healthy volunteers. (Kubota R, et al. IOVS 2009;50:ARVO E-Abstract 5009) A Phase II study is under way looking at ACU-4429 for patients with GA.
Fenretinide is a synthetic retinoid derivative that competes with retinol in the circulation by binding retinol-binding protein. The ensuing complex is small enough to be excreted through the kidneys, thereby decreasing the available pool of retinol for uptake at the RPE. Results of a Phase II trial investigating oral fenretinide for patients with GA showed reductions in lesion growth that correlated with reductions in retinol-binding protein. (Vogel R, et al. IOVS 2009;50:ARVO E-Abstract 4919) Results also showed that fenretinide decreased the incidence of choroidal neovascularization. Plans for a Phase III trial are under way.
Protecting Photoreceptors & RPE
Neuroprotective agents under investigation for dry AMD include ciliary neurotrophic factor (CNTF), brimonidine tartrate and tandosporine. All of these have been shown to decrease photoreceptor cell death in animal models of retinal degeneration. CNTF, a member of the IL-6 cytokine family, has been shown to retard cellular and functional losses not only in models of retinitis pigmentosa but other neurodegenerative diseases, as well. Investigators at Neurotech have developed an encapsulated cell implant with the ability to secrete CNTF within the vitreous cavity. Designated NT-501, the implant is approximately 5 mm in length and contains human RPE cells genetically engineered to produce CNTF at a constant rate. The outer wall of the implant is constructed of a semipermeable membrane with pores that allow CNTF to diffuse into the vitreous while protecting the RPE cells within from host-mediated immunologic attack. Preliminary results from a Phase II study looking at NT-501 for GA found stabilization of vision preceded by a statistically significant increase in retinal thickness.6 At one year, 96.3 percent of patients in the high-dose group lost fewer than three lines of vision, versus 75 percent of patients in the low-dose and sham-treatment group (p<0.078).
Brimonidine tartrate, an alpha-2 adrenergic receptor agonist, has been shown to reduce apoptosis in animal models of retinal injury, including ischemia, ocular hypertension, phototoxicity and partial optic nerve crush. A sustained-release brimonidine implant has been developed and is in clinical trials for patients with bilateral GA. Patients will be given either a 200 µg or 400 µg dose every six months with the fellow eye serving as a control. Outcome measures will include visual acuity, change in area of geographic atrophy, reading speed, and contrast sensitivity.7
Tandospirone is a topical, selective serotonin 1A agonist that has been shown to have neuroprotective effects in animal models. Studies have shown a dose-dependent protection of photoreceptors and RPE cells from severe photo-oxidative stress.8 A Phase III trial looking at tandospirone for patients with geographic atrophy has recently completed recruitment.
Reduction of toxic by-products and extracellular deposits is another source of promising research. The finding that drusen contain amyloid deposits has prompted investigators to compare the similarities between Alzheimer’s disease and other neurological diseases with dry AMD. Two drugs are being evaluated that have shown reduction of extracellular deposits, Copaxone and RN6G (Pfizer). Copaxone, or glatiramer acetate, is an FDA-approved drug for the treatment of multiple sclerosis and is given as a once daily subcutaneous injection. It has been found to suppress T-cells and down-regulate inflammatory cytokines. When the drug was evaluated for the treatment of Alzheimer’s disease it was found to reduce drusen.9 Though drusen reduction has been seen following laser treatment it was not associated with visual improvement. The hope is that the mechanism of drusen reduction by glatiramer acetate is different and that this may slow down visual loss. RN6G is an antibody against amyloid-β and has been shown in a mouse model to reduce amyloid-β deposits in the retina when administered systemically. Phase II trials are under way.
 |
Suppression of inflammation is another emerging treatment strategy for dry AMD. Strong evidence now exists that implicates the complement pathway as a key mechanism in the pathogenesis of AMD.10 Polymorphic variants in genes encoding complement factor H, complement component 3, and age-related maculopathy susceptibility 2 have been shown to significantly increase the risk of developing AMD. Histological analysis of patients with AMD has also revealed the presence of complement component 5 as well as the terminal membrane attack complex, C5b-9, within drusen.
A number of different complement inhibitors are currently under investigation for the treatment of AMD. One of the drugs is POT-4, a compastatin derivative that inhibits complement component 3. POT-4 reversibly binds complement component 3 and prevents its proteolytic activation to C3a and C3b, which eventually leads to the formation of the terminal membrane attack complex. POT-4 forms into an intravitreal gel at higher doses, which provides slow-release of the drug and requires less-frequent injections. A Phase I trial has already demonstrated safety and tolerability in humans as well as a reduction of leakage at higher doses in patients with wet AMD. A Phase II trial is under way.
Another complement inhibitor being investigated for AMD is eculizumab, a drug that has already been FDA-approved for the treatment of paroxysmal nocturnal hemoglobinuria. Eculizumab is a humanized IgG antibody administered intravenously that acts specifically against complement component 5. It is currently in an ongoing Phase II trial for the treatment of dry AMD. Eculizumab is particularly attractive in that it inhibits further downstream on the complement cascade than C3 inhibitors, thereby reducing inhibition of unwanted pathways. Other complement inhibitors include ARC-1905, FCFD4514S, JPE1375 and TA106.
In addition to the complement inhibitors, another promising anti-inflammatory drug being investigated for the treatment of dry AMD is the Iluvien implant, a nonbioerodible polyimide tube containing 180 µg of the corticosteroid fluocinolone acetonide. The implant is inserted via a 25-ga. intravitreal injector, which creates a self-sealing wound. A Phase II study is under way comparing two escalating doses with a control group in patients with GA.
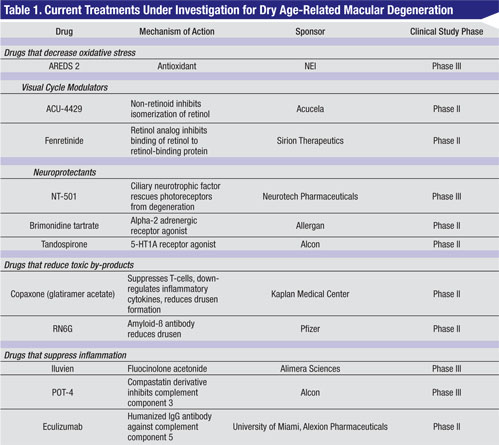
Other than AREDS vitamins, there is no other proven effective therapy for dry AMD. However, many trials are under way looking at a multitude of different treatments targeting different points along the pathways implicated in the pathogenesis of AMD (See Table 1).
Ultimately, it appears that the most effective treatment for AMD will likely be a combination of different drugs targeting the varying pathways that contribute to the development and progression of AMD.
Drs. Roe and Boyer practice at Retina-Vitreous Associates. Dr. Boyer is also in the Department of Ophthalmology, Keck School of Medicine, University of Southern California. Contact Dr. Roe at
roe.rick@gmail.com. Dr. Boyer is a consultant and scientific advisor for Alcon, Allergan, Eyetech, Genentech, Neurotech, Novartis, Pfizer and a scientific advisor with Regeneron.
1. Mata NL, Vogel R. Pharmacologic treatment of atrophic age-re-lated macular degeneration. Curr Opin Ophthalmol 2010;21:190-6.
2. Beatty S, Koh H, Phil M, et al. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 2000;45:115-34.
3. Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: An integrated survey of emerging treatment alternatives. Retina 2010;30:1350-67.
4. Meleth AD, Wong WT, Chew EY. Treatment for atrophic macular degeneration. Curr Opin Ophthalmol 2011;22:190-3.
5. Canter JA, Olson JM, Spencer K, et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS One 2008;3:e2091.
6. Zhang K, Hopkins JJ, Heier JS, et al. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci USA 2011;108:6241-5.
7. Safety and efficacy of brimonidine intravitreal implant in patients with geographic atrophy due to age-related macular degeneration (AMD). http://clinicaltrials.gov/ct2/show/NCT00658619
8. Collier RJ, Wang Y, Smith SS, et al. Complement deposition and microglial activation in the outer retina in light-induced retinopathy: Inhibition by a 5-HT1A agonist. Invest Ophthalmol Vis Sci 2011 Apr 5. [Epub ahead of print].
9. Landa G, Butovsky O, Shoshani J, et al. Weekly vaccination with Copaxone (glatiramer acetate) as a potential therapy for dry age-related macular degeneration. Curr Eye Res 2008;33:1011-3.
10. Charbel Issa P, Chong NV, Scholl HP. The significance of the complement system for the pathogenesis of age-related macular degeneration—current evidence and translation into clinical application. Graefe’s Arch Clin Exp Ophthalmol 2011;249:163-74.