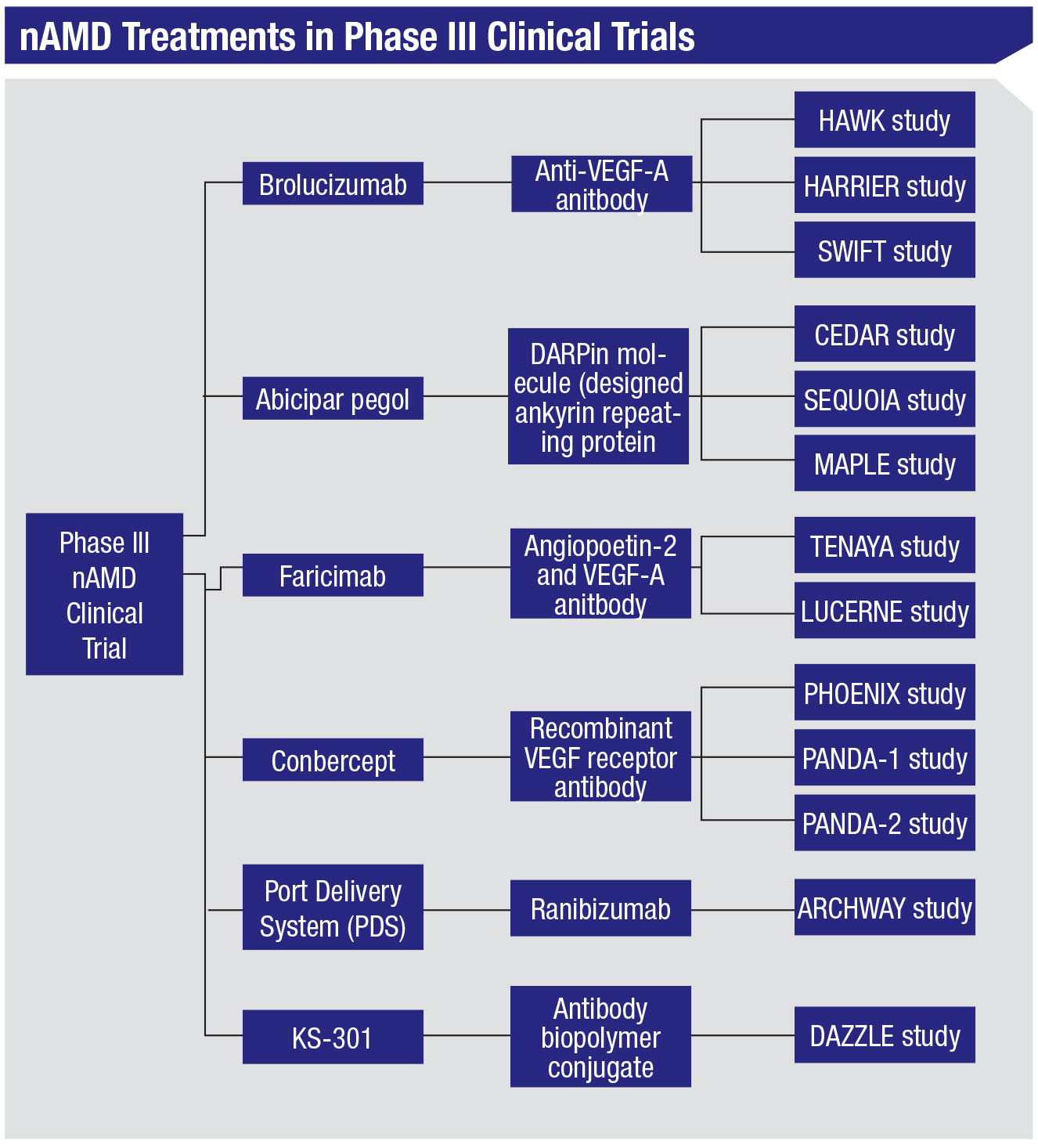
Though intravitreal anti-vascular endothelial growth factor agents are the mainstay of treatment for neovascular age-related macular degeneration, frequent and repeated injections are required in order to maintain the visual gains the drugs provide.1 This represents a substantial treatment burden to patients, with significant associated time and financial costs.2 Unfortunately, the need for frequent injections can often pose problems to treatment adherence and cause patients to be lost to follow-up.3 As a result, in recent years there’s been an increased focus on improving durability in novel therapeutics for nAMD in order to reduce this burden. In this article, we’ll review the current status of several nAMD medications that are now undergoing, or have recently completed, their Phase III clinical trials (Figure 1).4
Brolucizumab (Novartis)
Though brolucizumab was approved by the Food and Drug Administration in October 2019 and is already on the market,5 it’s still undergoing Phase III and IV clinical trials to explore its safety and efficacy. Brolucizumab is an anti-VEGF-A antibody grafted to a human single-chain antibody fragment. Use of the single chain fragment, the smallest antibody unit, allows for a greater dose of anti-VEGF per unit of volume.6,7 This allows higher concentration in the vitreous and more penetrability of the retina and RPE. Furthermore, a higher dose per unit of anti-VEGF can allow for slower clearance from the eye, and multiple studies have shown that brolucizumab can potentially allow for extended dosing schedules. In the first Phase I/II human trial of brolucizumab, the median time for re-dosing anti-VEGF after receiving brolucizumab 6 mg was 30 days longer than the median time required for re-dosing anti-VEGF after receiving ranibizumab.8 The Phase II study OSPREY, which found non-inferiority of brolucizumab 6 mg to aflibercept 2 mg dosed at eight weeks, laid the foundation for 12-week dosing.7
The Phase III trials that paved the way for FDA approval of brolucizumab were HAWK and HARRIER. HAWK randomized 1,082 patients to three groups in a 1:1:1 fashion: brolucizumab 3 mg or 6 mg or aflibercept 2 mg. HARRIER randomized 743 patients equally to either brolucizumab 6 mg or aflibercept 2 mg. In both trials, all groups received a loading dose of three monthly treatments. Additionally, aflibercept was dosed every eight weeks, whereas brolucizumab was initially dosed every 12 weeks. If disease activity was detected by pre-specified functional and structural measures, those in the brolucizumab group permanently dropped down to eight-week dosing. At Week 48, the primary endpoint of each trial was achieved, with brolucizumab demonstrating noninferiority to aflibercept in BCVA change from baseline. In each group there was an improvement in visual acuity by about six to seven ETDRS letters over 48 weeks. Also, 56 percent of patients in HAWK and 51 percent of patients in HARRIER were able to stay on 12-week dosing of brolucizumab. Patients on brolucizumab also had a greater significant reduction in central subfield thickness as compared to those on aflibercept.6
Although the overall safety profiles between brolucizumab and aflibercept were initially thought to be similar,6 brolucizumab has since come under scrutiny for reports of vision loss secondary to severe noninfectious uveitis and occlusive retinal vasculitis.9 The American Society of Retina Specialists and Novartis have been diligent about continued surveillance, and Novartis has launched a new website that provides updates of significant adverse events. As of June 2020, the incidence of retinal vasculitis and/or occlusion is 8.65 per 10,000 injections. Novartis and the ASRS have not recommended against use of the medication but encourage physician vigilance in looking for signs of inflammation.10,11
Several other Phase IIIb trials are under way and remain active. One recent addition is the SWIFT study, a new single-arm trial that will evaluate patients to determine the effect of brolucizumab using a treat-and-extend regimen in patients with nAMD. Patients receive three monthly doses of brolucizumab, and then are extended by four weeks up to a 16-week interval. The primary outcome is the proportion of patients with no disease activity at week 48.12
 |
Abicipar pegol (AbbVie/Allergan & Molecular Partners)
Abicipar pegol is a designed ankyrin repeating protein (DARPin) molecule. It’s a recombinant protein that binds all isoforms of VEGF-A. Its small size allows for easily achievable higher molar concentration, leading to higher vitreous concentration and better tissue penetration. Furthermore, since its molecules have high binding affinity and good stability it has the potential to be quite durable.13
In October 2019, Allergan and Molecular Partners announced their two-year data from two identical Phase IIIa trials, CEDAR and SEQUOIA, evaluating the efficacy and safety of abicipar as compared to ranibizumab. Patients with nAMD were assigned to receive abicipar 2 mg every eight weeks after a loading dose of three monthly injections; abicipar 2 mg every 12 weeks after a loading dose of two monthly injections followed by an injection after eight weeks; or ranibizumab 0.5 mg monthly.14,15 The primary outcome for each of the studies was the proportion of patients with stable vision, defined as a change in vision by no more than 15 ETDRS letters.
At 52 weeks, abicipar met the noninferiority criteria for the primary outcome in both the q8 and q12 week dosing arm, with 93 percent and 90 percent of patients maintaining baseline vision in pooled results from both studies. Secondary endpoints, such as mean best-corrected visual acuity, were maintained from year one to year two across all arms, and central subfield thickness decreased at year one and again at year two across all arms.16
In year one there were initial concerns about high rates of new-onset intraocular inflammation: 15.4 percent in the abicipar every-eight-week group and 15.1 percent in the abicipar every-12-week group, as compared to none in the ranibizumab group.17 New intraocular inflammation declined in the second year of the study, with a pooled rate of 1.9 percent in both abicipar arms and 1 percent in the ranibizumab arm.16 Because of these initial safety concerns, the MAPLE study, a 28-week safety evaluation, was performed to determine the rates of adverse events in 128 patients after the manufacturing process was changed. The study had a single arm of nAMD patients treated with a loading dose of three monthly injections followed by injections every eight weeks. The study found a lower rate, 8.9 percent, of intraocular inflammation than CEDAR and SEQUOIA. The incidence of severe intraocular inflammation was 1.6 percent.17
In late June of 2020, the FDA didn’t approve abicipar pegol for use, citing that the rates of observed intraocular inflammation resulted in an unfavorable benefit-risk ratio in the treatment of patients with neovascular AMD. AbbVie, which has completed its acquisition of Allergan, has yet to formally announce any plans regarding the future of the molecule.18
Faricimab (Roche/Genentech)
Faricimab is a novel antibody that’s bi-specific, inhibiting both angiopoetin-2 and VEGF-A. This bi-specificity is theorized to lead to sustained efficacy. The 52-week results of the Phase II STAIRWAY study were previewed at the 2018 American Academy of Ophthalmology’s meeting and the 2020 virtual meeting of the Association for Research in Vision and Ophthalmology. The study had three arms, evaluating 76 patients with nAMD who were randomized to either faricimab 6 mg every 12 or 16 weeks (after initial monthly loading doses) or ranibizumab 0.5 mg every four weeks.19,20 At week 24, 65 percent of patients treated with faricimab had no active disease. People dosed every 16 weeks had a mean best-corrected visual acuity improvement of 11.4 mean letters from baseline at week 52. At 52 weeks, faricimab every 12 weeks provided a gain of 10.1 letters vs. 9.6 letters with ranibizumab.19, 21 Each arm of the trial had a similar proportion of greater than 15 letters gained and fewer than 15 letters lost. There were no appreciable differences in adverse events or other safety outcomes.21 Following the success of the Phase II trial, two identical, multicenter Phase III trials, TENAYA and LUCERNE, are under way to further evaluate safety and efficacy. The studies have enrolled 1,280 participants, randomized to faricimab every 16 weeks—with the possibility of decreasing the interval to every 12 or eight weeks—or aflibercept every eight weeks.22 The primary outcome measure is the average change of BCVA from baseline to week 48 in the patients. The targeted completion date is the fall of 2022.23,24

|
Conbercept (Chengdu Kanghong Biotech)
Conbercept is a recombinant VEGF receptor antibody, thought to have improved stability over aflibercept because of its combination of the second immunoglobulin domain of VEGFR1 and the third and fourth immunoglobulin domain of VEGFR2.25 The PHOENIX trial was a 12-month, randomized, Phase III trial of conbercept performed at nine locations throughout China that enrolled 114 patients. The study evaluated intravitreal injections of conbercept 0.5 mg with a three-month loading dose followed by quarterly injections. The control group received a delayed regimen: sham injections (as there were no previously approved anti-VEGF agents in China) monthly for three months, followed by three monthly injections, followed by quarterly injections up until 12 months. The primary outcome was mean change in BCVA at three months.
At this three month primary endpoint, there was a statistically significant difference in the change in BCVA from baseline, with a gain of 9.2 letters in the conbercept group and a gain of only 2.02 letters in the sham group. At one year, and after both groups received the study medication, there was improved BCVA from baseline, with the conbercept group showing an increase in BCVA by 9.98 letters and the (initial) sham group showing a gain of 8.81. This difference wasn’t statistically significant at the 12-month endpoint. Though there were no significant safety concerns during the study, PHOENIX wasn’t adequately powered to detect all adverse events.26
After the National Medical Products Administration’s (the Chinese equivalent to the U.S. Food and Drug Administration) approval of conbercept in 2013, the U.S. FDA expedited conbercept to undergo Phase III clinical testing without repeat Phase I or II trials in the United States. Beginning in 2018, two parallel Phase III studies, PANDA-1 and PANDA-2, were tasked to evaluate conbercept globally in treatment-naïve nAMD patients. Both trials will evaluate the safety and efficacy of conbercept 0.5 mg at eight-week intervals and conbercept 1 mg at 12-week intervals, as compared to aflibercept 2 mg at eight-week intervals (control arm), after each arm receives three monthly loading doses. The combined enrollment target is 1,140 patients, randomized evenly between the three arms. The primary endpoint is the change in BCVA at 36 weeks from baseline. Secondary outcomes include evaluating both functional and structural improvements. The main difference between PANDA-1 and PANDA-2 is that the former follows continuous dosing at the above intervals through week 92. At week 40, PANDA-2 adopts pro re nata injections up to a cap of 16-week intervals. Each study is expected to finish by 2022.27,28
Port Delivery System (Roche/Genentech)
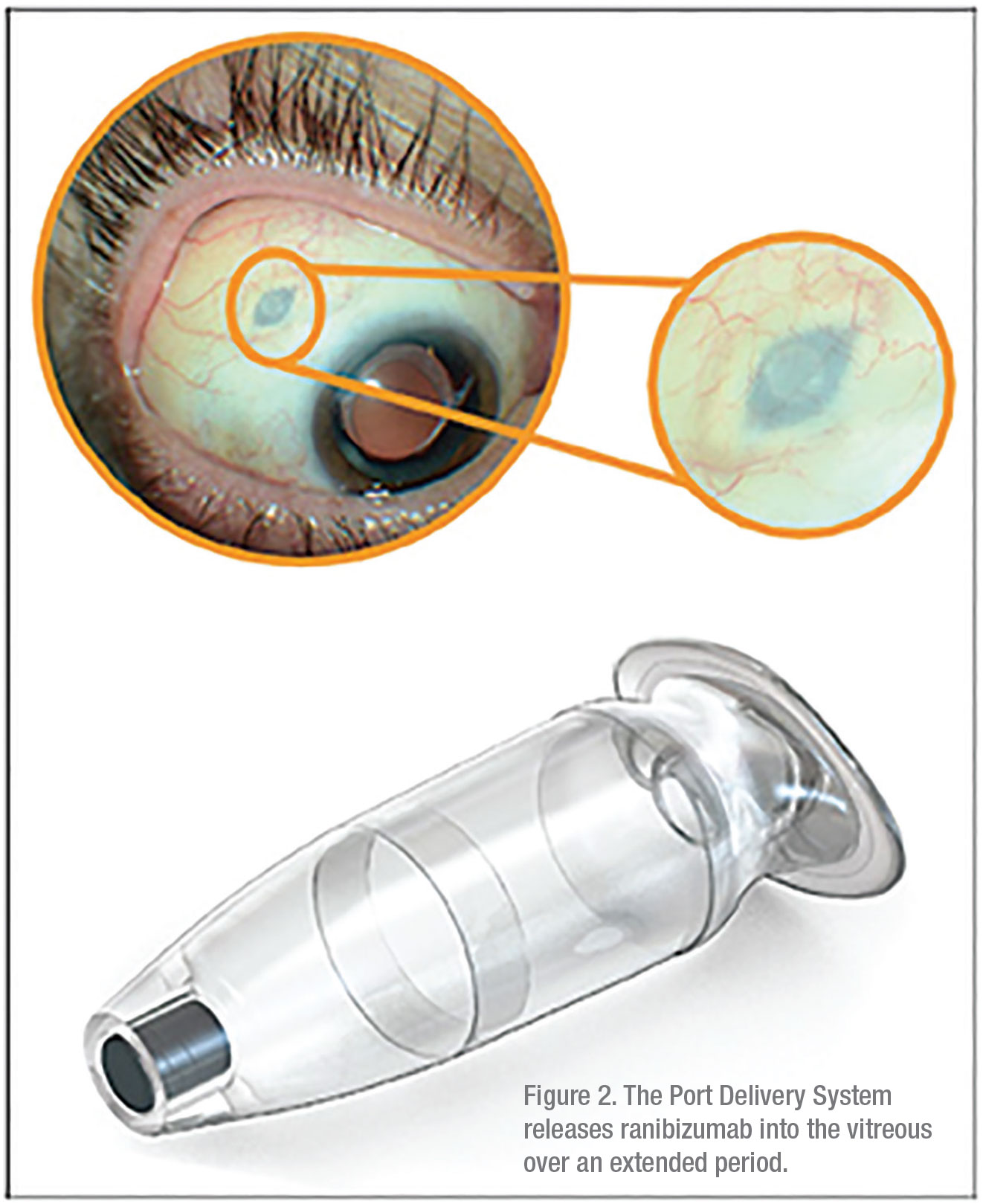
While other Phase III trials discussed thus far have focused on novel therapeutics, the port delivery system, or PDS, focuses on a novel delivery system. The PDS is an implant designed to administer a highly concentrated form of ranibizumab over an extended period. After the device is surgically implanted through an incision in the sclera at the pars plana (Figure 2), it continuously releases ranibizumab into the vitreous via passive diffusion. The self-sealing device can then be refilled in the office via injection through the conjunctiva.
The Phase II randomized controlled clinical trial, LADDER, demonstrated initial visual and anatomic success with the PDS. The trial randomized 220 nAMD patients in a 3:3:3:2 ratio to four different arms: ranibizumab 10 mg/ml, ranibizumab 40 mg/ml, or ranibizumab 100 mg/ml using the PDS or standard monthly intravitreal ranibizumab 0.5-mg injections. The PDS with 100 mg/ml of ranibizumab demonstrated the longest median time until refill, at 15 months, compared to 8.7 and 13 months in the 10 mg/ml and 40 mg/ml groups.
In terms of visual outcomes, at nine months, the BCVA change from baseline was a loss of ETDRS letters in the PDS 10 mg/ml and 40 mg/ml. By comparison, there was a gain of 5.1 and 3.9 ETDRS letters in the PDS 100 mg/ml and monthly intravitreal ranibizumab 0.5 mg arms, respectively. Mean CFT change from baseline was similar at nine months between the 100 mg/ml PDS and monthly ranibizumab groups. The primary adverse event was vitreous hemorrhage, which occurred at a rate of 4.5 percent.29
Given the success of the Phase II trial, the PDS is undergoing an additional Phase III study, ARCHWAY, which began in 2018 and is estimated to finish in 2022. The study randomized patients to PDS 100 mg/ml at baseline with fixed refill intervals of 24 weeks. The control arm is monthly intravitreal injections of 0.5-mg ranibizumab. The primary endpoint, the change in BCVA, will be determined by averaging the BCVA scores of weeks 36 and 40 and comparing that value to the baseline score. Secondary outcomes include evaluating the change in central subfoveal thickness from baseline, and rates of adverse events.30
In addition, PORTAL is an extension Phase III study designed to look at long-term safety, both ocular and systemic, of the PDS 100 mg/ml in patients who have completed the ARCHWAY or LADDER trial. Those who received monthly intravitreal injections were also eligible for PDS implantation. The study plans to evaluate patients with the PDS receiving in-office refills every 24 weeks for 144 weeks.31
 |
KSI-301 (Kodiak Sciences)
KSI-301 is an anti-VEGF IgG1 antibody linked to a high molecular weight biopolymer. The large molecule allows for high molar concentrations of anti-VEGF-A binding capability, which is posited to extend durability. In Phase Ib, 35 patients with treatment-naïve nAMD were given 2.5- or 5-mg doses of KSI-301. At 16 weeks, best-corrected visual acuity improved by a mean of 5.4 letters, and after three monthly loading doses, 80 percent were able to extend their treatment intervals to as long as four months before the next injection.32 Its success in Phase Ib has led to a pivotal Phase IIb/III trial called DAZZLE. Estimating an enrollment of 550 participants, DAZZLE will randomize patients to receive KSI-301 5 mg or sham at 12-, 16- and 20-week intervals. A comparator arm will enroll participants to receive aflibercept 2 mg or sham for three monthly loading doses followed by every-eight-week injections. Primary outcome is the mean change in best-corrected visual acuity at a year.33
In conclusion, with a focus on enhanced duration, the current Phase III clinical trials in nAMD provide new hope for expanding our therapeutic armamentarium. Taken together, these new therapies could provide a more comprehensive approach to caring for patients with nAMD. REVIEW
Dr. Soares is a vitreoretinal Fellow at Mid Atlantic Retina/Wills Eye Hospital. Dr. Mahmoudzadeh is a postdoctoral retina research Fellow at Wills Eye. Dr. Cohen is a member of the Retina Service of Wills Eye, and is an assistant professor of ophthalmology at the Sidney Kimmel Medical College at Thomas Jefferson University.
Dr. Cohen is a consultant for Allergan.
1. Preferred Practice Patterns: Age-Related Macular Degeneration. American Academy of Ophthalmology; 2019. Available at: https://www.aao.org/preferred-practice-pattern/age-related-macular-degeneration-ppp.
2. Brown GC, Brown MM, Rapuano S, Boyer D. Cost-utility analysis of VEGF inhibitors for treating neovascular age-related macular degeneration. Amer J Ophthalmol 2020;218:225–241.
3. Obeid A, Gao X, Ali FS, et al. Loss to follow-up among patients with neovascular age-related macular degeneration who received intravitreal anti-vascular endothelial growth factor injections. JAMA Ophthalmol 2018;136:1251–1259.
4. Ammar MJ, Hsu J, Chiang A, et al. Age-related macular degeneration therapy: A review. Curr Opin Ophthalmol 2020;31:215–221.
5. Drug Approval Package: BEOVU (brolucizumab-dbll). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761125_Orig1_toc.cfm [Accessed June 7, 2020].
6. Dugel PU, Koh A, Ogura Y, et al. HAWK and HARRIER: Phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology 2020;127:72–84.
7. Dugel PU, Jaffe GJ, Sallstig P, et al. Brolucizumab versus aflibercept in participants with neovascular age-related macular degeneration: A randomized trial. Ophthalmology 2017;124:1296–1304.
8. Holz FG, Dugel PU, Weissgerber G, et al. Single-chain antibody fragment VEGF inhibitor rth258 for neovascular age-related macular degeneration: A randomized controlled study. Ophthalmology 2016;123:1080–1089.
9. Baumal CR, Spaide RF, Vajzovic L, et al. Retinal vasculitis and intraocular inflammation after intravitreal injection of brolucizumab. Ophthalmology 2020;127:10:1345-1359.
10. Hahn P, Arevalo JF, Blinder K, et al. Occlusive retinal vasculitis following intravitreal brolucizumab: An ASRS Research and Safety in Therapeutics (ReST) committee report. ASRS Research and Safety in Therapeutics (ReST) Committee; 2020.
11. Safety of Beovu (brolucizumab). Available at: https://www.brolucizumab.info/ [Accessed June 25, 2020].
12. Novartis Pharmaceuticals. A one-year, single-arm, open-label, multicenter study assessing the effect of brolucizumab on disease control in adult patients with suboptimal anatomically controlled neovascular age-related macular degeneration (SWIFT). Available at: https://clinicaltrials.gov/ct2/show/NCT04264819 [Accessed June 4, 2020].
13. Rodrigues GA, Mason M, Christie L-A, et al. Functional characterization of abicipar-pegol, an anti-VEGF DARPin therapeutic that potently inhibits angiogenesis and vascular permeability. Invest Ophthalmol Vis Sci 2018;59:5836–5846.
14. Safety and efficacy of abicipar pegol in patients with neovascular age-related macular degeneration. ClinicalTrials.gov identifier: NCT02462486. https://clinicaltrials.gov/ct2/show/NCT02462486. Accessed 6/8/2020.
15. A safety and efficacy study of abicipar pegol in patients with neovascular age-related macular degeneration (CEDAR). ClinicalTrials.gov identifier: NCT02462928. https://clinicaltrials.gov/ct2/show/NCT02462928. Accessed 6/8/2020.
16. Allergan and Molecular Partners news release. Available at: https://www.molecularpartners.com/allergan-and-molecular-partners-present-late-breaking-data-from-phase-3-studies-of-investigational-abicipar-pegol-in-neovascular-wet-age-related-macular-degeneration. [Accessed June 7, 2020].
17. Allergan and Molecular Partners Announce Topline Safety Results from MAPLE study of Abicipar pegol – Molecular Partners. Available at: https://www.molecularpartners.com/allergan-and-molecular-partners-announce-topline-safety-results-from-maple-study-of-abicipar-pegol/ [Accessed June 9, 2020].
18. Allergan/AbbVie news release. Available at: https://news.abbvie.com/news/press-releases/allergan-an-abbvie-company-and-molecular-partners-receive-complete-response-letter-from-fda-on-biologics-license-application-for-abicipar-pegol.htm [Accessed September 8, 2020].
19. STAIRWAY Analysis. HCPLive. Available at: https://www.mdmag.com/conference-coverage/arvo-2020/stairway-analysis-effects-faricimab-use-for-namd [Accessed June 11, 2020].
20. Study to evaluate faricimab (RO6867461; RG7716) for extended durability in the treatment of neovascular age related macular degeneration (nAMD). ClinicalTrials.gov. Available at: https://clinicaltrials.gov/ct2/show/record/NCT03038880 [Accessed June 11, 2020].
21. Genentech news release. Available at: https://www.gene.com/media/press-releases/14762/2018-10-27/new-stairway-study-data-shows-potential. [Accessed June 11, 2020].
22. Roche and Genentech take global macular degeneration step. Pharma Times. Available at: http://www.pharmatimes.com/news/roche_and_genentech_take_global_macular_degeneration_step_1280945 [Accessed October 1, 2020].
23. Clinicaltrials.gov. A Phase III, multicenter, randomized, double-masked, active comparator-controlled study to evaluate the efficacy and safety of faricimab in patients with neovascular age-related macular degeneration (TENAYA). Available at: https://clinicaltrials.gov/ct2/show/NCT03823287 [Accessed June 8, 2020].
24. Clinicaltrials.gov. A Phase III, multicenter, randomized, double-masked, active comparator-controlled study to evaluate the efficacy and safety of faricimab in patients with neovascular age-related macular degeneration (LUCERNE). Available at: https://clinicaltrials.gov/ct2/show/NCT03823300 [Accessed June 8, 2020].
25. Zhang M, Yu D, Yang C, et al. The pharmacology study of a new recombinant human VEGF receptor-fc fusion protein on experimental choroidal neovascularization. Pharm Res 2009;26:204–210.
26. Liu K, Song Y, Xu G, et al. Conbercept for treatment of neovascular age-related macular degeneration: Results of the randomized phase 3 PHOENIX Study. Am J Ophthalmol 2019;197:156–167.
27. PANDA-1 study, clinicaltrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03577899 [Accessed June 29, 2020].
28. PANDA-2 study, clinicaltrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03630952 [Accessed June 29, 2020].
29. Campochiaro PA, Marcus DM, Awh CC, et al. The Port Delivery System with ranibizumab for neovascular age-related macular degeneration: Results from the randomized phase 2 ladder clinical trial. Ophthalmology 2019;126:1141–1154.
30. Archway study, clinicaltrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03677934.
31. Portal study, clinicaltrials.gov. Available at: https://clinicaltrials.gov/ct2/show/NCT03683251 [Accessed June 22, 2020].
32. Wykoff CC. Extended durability in exudative retinal diseases using the novel intravitreal anti-VEGF antibody biopolymer conjugate KSI-301: Results from the Phase 1b study in patients with AMD, DME and RVO. Presented at the 2019 American Academy of Ophthalmology Retina Subspecialty Day; October 11, 2019; San Francisco.
33. Dazzle study, clinicaltrials.gov. Kodiak Sciences Inc. Available at: https://clinicaltrials.gov/ct2/show/NCT04049266 [Accessed September 10, 2020].
33. Kodiak Sciences Inc. A Phase IIb/III, prospective, randomized, double-masked, active comparator-controlled, multi-center study to investigate the efficacy and safety of repeated intravitreal administration of KSI-301 in subjects with neovascular (wet) age-related macular degeneration. clinicaltrials.gov; 2020. Available at: https://clinicaltrials.gov/ct2/show/NCT04049266 [Accessed September 10, 2020].