Inherited retinal diseases (IRDs) are typically caused by single-gene mutations and are historically classified by clinical features, imaging and electroretinography. The advent of low-cost genetic testing now enables more precise and granular classification of IRDs, which can also guide prognosis and management if the causative mutation is identified.1 More than 300 such diseases have been identified, and cumulatively they affect approximately 200,000 people in the United States and 4.5 million people worldwide.2, 3 On the treatment side, the field of gene therapy has advanced significantly in recent years, with many clinical trials in various stages under way, including 47 that are currently recruiting, 25 beginning soon and 72 that have been completed.4
Here, we survey recent progress in gene therapies for several classes of IRDs, including photoreceptor disorders such as retinitis pigmentosa and Leber congenital amaurosis, macular dystrophies including Stargardt disease and X-linked retinoschisis, as well as choroidal dystrophies such as choroideremia.
An Overview of Gene Therapy
The term gene therapy commonly refers to gene replacement, where a normal, functional copy of a gene is introduced by way of a delivery vector to replace a mutated gene in a targeted population of cells. This approach is commonly used for recessive IRDs where neither of the two mutated alleles can produce functional gene products. In contrast, dominant IRDs may require therapies that inactivate the mutant protein or gene at the DNA level using gene-editing technologies like CRISPR.
The challenges of developing gene therapies vary with different IRDs, based on differences in the therapeutic transgene, the intended target cell type and clinical course, among many other factors. Viral vectors such as adenovirus, adeno-associated virus (AAV) and lentivirus differ in their gene-carrying capacity, cellular tropism, immunogenicity and mutagenicity. Different routes of administration, including intravitreal, subretinal and suprachoroidal, provide different biodistribution and may differentially trigger host immune responses to viral particles in different compartments surrounding ocular barriers (See Figure 1).5 Finally, the clinical course may dictate the window of opportunity for which gene therapy can be effective, before photoreceptor atrophy leads to irreversible blindness.
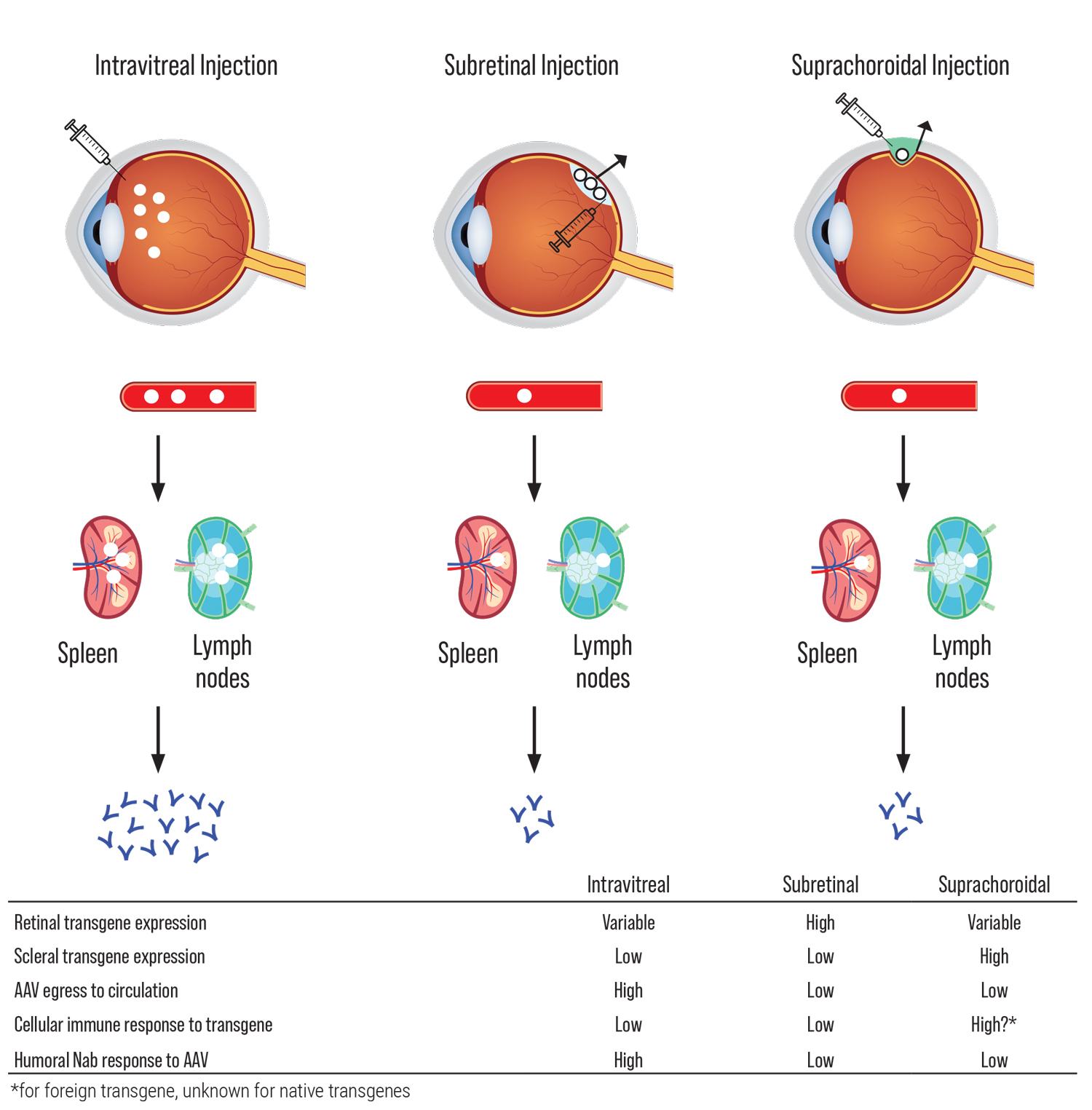 |
| Figure 1. Routes of viral vector delivery. Effects of different routes of viral vector delivery on transgene expression, egress to circulation and immune response. AAV indicates “adeno-associated virus”; Nab, “neutralizing antibodies.”6 (Click image to enlarge.) |
Leber Congenital Amaurosis
The first major success for retinal gene therapy was in the treatment of Type 2 LCA, an autosomal recessive IRD that occurs in one in 80,000 births, and is associated with mutations in the GUCY2D, CEP290 and RPE65 genes.7 RPE65 is involved in the production of 11-cis-retinal during phototransduction, and accounts for 5 to 10 percent of LCA cases. Voretigene neparvovec-rzyl (Luxturna, Spark Therapeutics, Philadelphia) is an AAV2 that delivers RPE65 via subretinal injection which demonstrated safety and benefit in Phase III studies and was approved by the FDA in 2017 for patients with biallelic RPE65-mediated IRDs.8, 9
Given the challenge of measuring improvements in functional vision in IRD patients, where visual acuity alone cannot suffice, a novel ambulatory navigation maze was devised as an end point in addition to light sensitivity and visual fields. Recent studies suggest that long-term functional improvements persist to at least three to four years after gene augmentation with VN; of note, no deleterious immune responses were observed.8 However, 18 eyes of 10 patients who underwent subretinal VN were recently noted to develop perifoveal chorioretinal atrophy, identified around five months after treatment and persisting for at least a year during early follow-up.10 This unexpected outcome resulted in some patients experiencing a scotoma, but very few had significant changes in visual acuity or other functional measures.
Another follow-up study followed 77 eyes of 41 patients and found that central foveal thickness decreased slightly in both children and adults, as the fovea was detached by VN in 62 eyes (81 percent). There was no statistically significant vision change for the adults, whereas there was a trend of improvement for children that reached statistical significance at some time points. At the last follow-up, 29 percent of the pediatric eyes improved by at least two lines.11
In 2022, a study of 27 eyes of 14 patients examined postoperative complications and longitudinal changes in photoreceptor function. The most common postoperative issues included elevation in intraocular pressure (59 percent), persistent intraocular inflammation (15 percent), and vitreous opacities (26 percent) that resolved over months, providing longitudinal real-world evidence of VN safety and efficacy consistent with the original clinical trial results.12
Type 10 LCA is caused by mutations in the CEP290 gene that result in a splicing error in the mRNA transcript of a protein which forms the primary cilium and plays an important role in photoreceptors. In October 2019, an antisense oligonucleotide designed to correct the splicing error was investigated in a Phase I/II clinical trial of 10 subjects in whom vision improved by several lines at three months, and the improvements in visual acuity were retained after six months with a second dose.13
Separately, in March 2020, the first in-human ophthalmic application of in vivo gene editing using a CRISPR-Cas system commenced to evaluate AGN-151587 (EDIT-101, Allergan; NCT03872479) delivered via subretinal injection in 18 patients. Early results have demonstrated both safety and efficacy in the first cohort, and the trial is on target to be completed in 2024.14
Choroideremia
Choroideremia (CHM) is an X-linked recessive IRD that occurs in one in 50,000 males, and presents with night blindness and gradual vision loss beginning in childhood. The CHM gene encodes the Rab escort protein 1 (REP1) essential for intracellular vesicular transport, and loss of function results in cell death and the gradual deterioration of the retinal pigment epithelium, photoreceptors and the choroid.15
In Phase I/II trials, subretinal AAV2-REP1 improved visual acuity in some patients, although there were a few cases of adverse events. Among six patients, there were two cases of retinal hole over non-functional retina16 and one case of localized intraretinal immune response.17 Sequential bilateral treatment using AAV2-REP1 was tested in the GEMINI open-label Phase II trial (NCT03507686) from Biogen (Cambridge, Massachusetts), and it appeared safe. However, the pivotal Phase III STAR trial which randomized one eye per subject to low dose, high dose or control groups (NCT03496012) didn’t meet the primary endpoint of proportion of participants with a ≥ 15 ETDRS letter improvement from baseline at month 12, although the safety results were consistent with previous studies. Recently, Spark Therapeutics initiated a Phase I/II study of unilateral subretinal administration of the AAV2-hCHM vector in choroideremia subjects with BCVA >20/200 in the study eye (NCT02341807). More recently, using adaptive optics, one group showed that the cone photoreceptor mosaic resettled on the RPE following resolution of the subretinal bleb at one month post-injection, remaining intact in eight of nine study participants without widespread cone loss across the retained area of central retina targeted by the retinal detachment, which suggests that cone photoreceptors don’t drop out as a consequence of mechanical or acute inflammatory changes in response to subretinal AAV2-hCHM.18
Stargardt Disease
As the most common macular dystrophy worldwide, with a prevalence of one in 10,000 people, Stargardt disease is an autosomal recessive disease that results from loss-of function mutations in the ABCA4 gene-encoding ATP-binding cassette A4 transporter. This protein clears toxic lipofuscin-component A2E from photoreceptors; its absence leads to progressive retinal degeneration with characteristic light-yellow pisciform flecks that’s accompanied by a sharp initial decline in central vision followed by a slow progressive decline. Visual acuity typically remains relatively preserved for several decades, rarely declining beyond 20/400. Nonsense mutations cause early onset disease in childhood with more severe atrophy, whereas missense variants are usually adult-onset, often sparing the fovea.19
Several novel pharmacologic therapies are currently being developed to decrease A2E formation in the visual cycle.19 With its high prevalence and broad phenotypic spectrum, ABCA4 is also an attractive target for gene therapy. Given the size of the ABCA4 gene (6.8kb), lentiviral or nanoparticle vectors are needed deliver the payload. A Phase I/II clinical trial (NCT01367444) investigating subretinal lentiviral delivery of ABCA4 using SAR422459 from Oxford Biomedica demonstrated positive early safety data, but was terminated prematurely due to loss of sponsorship. A long-term follow-up study of patients who received this treatment (NCT01736592) is currently ongoing.20
X-Linked Retinitis Pigmentosa
Retinitis pigmentosa is characterized by progressive vision loss due to abnormalities of photoreceptor cells or retinal pigment epithelial cells, with a prevalence of 1 in 4,000. While three genes—RHO, USH2A, and RPGR—account for about 30 percent of all RP cases, a total of 87 individual disease-causing genes have been identified to date. Although RP can be inherited in all three major Mendelian patterns, X-linked RP generally has an earlier onset and worse prognosis.
Due to the large number of causative genes and phenotypic variance in RP, clinical findings, onset and progression may differ considerably. Typically, the disease begins with damage and loss of rod cells, leading to nyctalopia, defective dark adaptation and peripheral visual field loss, which is then followed by eventual secondary loss of cones, causing central visual loss.21
For X-linked RP caused by mutations in RPGR, a recombinant AAV2/5 vector MGT009 (Mereira/Janssen) has been designed to subretinally deliver functional copies of the gene in males with XLRP. In a Phase I/II dose escalation trial, four patients in the intermediate-dose cohort achieved clinically-meaningful improvements in visual field progression at 12 months, while those in the low- and intermediate-dose cohorts (n=6) also achieved significant improvements in their vision-guided mobility maze evaluation. There were no reports of dose-limiting events, although signs of inflammation were observed in two of three patients in the high-dose cohort, who were successfully managed with steroids.22 Plans to proceed with a Phase III trial are under way (NCT04671433).
Similarly, AGTC-501 (AGTC) is a recombinant AAV2 administered by subretinal injection for RPGR-related XLRP patients. Preliminary results showed that the therapy was well-tolerated across a wide dose range with minimal adverse effects. At 12 months, four out of eight patients were considered responders, and a planned Phase II/III will randomize 63 participants to compare two doses in the future.
X-Linked Retinoschisis
XLRS has a prevalence of 1 in 5,000 to 20,000, and is associated with mutations of the RS1 gene that encodes the membrane protein retinoschisin, involved in retinal cell layer organization and cell adhesion, as well as ion-channel mediated fluid balance.23 Male patients typically present within the first two decades of life with predominantly central vision loss. The macular schisis creates the appearance of radial folds emanating from the fovea, and may eventually extend to the peripheral retina. Visual acuity is decreased in XLRS, but may remain relatively stable for long periods of time. There’s no current treatment for XLRS, and management is focused on preserving vision and addressing complications such as recurrent retinal detachment and vitreous hemorrhage.
Intravitreal injections are the preferred approach for gene therapy delivery, since the retina is predisposed to retinal detachment with subretinal injections leading to decreased structural integrity. AAV-encoding RS1 has been proven safe and effective in preclinical trials using RS1-knockout mice and macaques.24, 25 Two ongoing Phase I/II clinical trials are investigating the safety of intravitreal AAV8 (NCT02317887) and AAV2 (NCT02416622). Early data suggest that eyes treated with intravitreal AAV8 exhibit concerning signs of inflammation, possibly due to a baseline proinflammatory state in XLRS.26 Advances in distinguishing the phenotypic variability of this disease may help improve trial design and timing of interventions. For example, measuring the integrity or length of photoreceptor outer segments on SD-OCT may help identify the optimal candidates for treatment.27
In conclusion, gene therapy can be a promising approach to treating IRDs, but challenges remain in vector design, mode of delivery and host immunity. New generations of AAV enable better penetration into the retina and potentially lower immunogenicity. Suprachoroidal injections using microneedles enable in-office delivery of gene therapies without the need for invasive vitreoretinal surgery. However, different modes of AAV delivery may elicit differential host immune responses that can trigger intraocular inflammation, causing permanent damage if not properly managed.
While much of the research to date has focused on gene augmentation, other emerging genetic therapies include RNA interference (RNAi) using siRNA or antisense oligonucleotide therapy, CRISPR/Cas9-based gene editing and base editing, and translational readthrough-inducing drugs. With large numbers of clinical trials under way, more advances in gene therapy technology are needed to guide a safe, steady path forward in this exciting new area of therapy.
Dr. Yiu is an associate professor at the University of California, Davis. He receives research support from Clearside, Iridex and Genentech. He consults for Abbvie, Adverum, Alimera, Anlong, Bausch & Lomb, Clearside, Endogena, Genentech, Gyroscope, Intergalactic, Iridex, NGM Bio, Regeneron, Thea, Topcon and Zeiss. Dr. Gupta is a PGY3 Ophthalmology Resident at Geisinger Health System. He has no disclosures.
This article has no commercial sponsorship.
1. Pulido JS, Procopio R, Davila HJ, Bello N, Ku C, Pennesi ME, Yang P, Nagiel A, Mahroo OA, Aleman TS et al. Inherited retinal disease panels—caveat emptor—truly know your inherited retinal disease panel. Retina 2022;42:1:1-3.
2. Benati D, Patrizi C, Recchia A: Gene editing prospects for treating inherited retinal diseases. J Med Genet 2020;57:7:437-444.
3. RetNet. https://sph.uth.edu/retnet/disease.htm. Accessed March 28, 2022.
4. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/results?term=inherited+Retinal&Search=Clear&age%20y=&gndr=&rsit=. Accessed March 28, 2022.
5. Yiu G, Chung SH, Mollhoff IN, Nguyen UT, Thomasy SM, Yoo J, Taraborelli D, Noronha G. Suprachoroidal and subretinal injections of AAV using transscleral microneedles for retinal gene delivery in nonhuman primates. Mol Ther Methods Clin Dev 2020;16:179-191.
6. Mehta N, Robbins DA, Yiu G. Ocular inflammation and treatment emergent adverse events in retinal gene therapy. Int Ophthalmol Clin 2021;61:3:151-177.
7. Tsang SH, Sharma T. Leber congenital amaurosis. Adv Exp Med Biol 2018;1085:131-137.
8. Maguire AM, Russell S, Chung DC, Yu ZF, Tillman A, Drack AV, Simonelli F, Leroy BP, Reape KZ, High KA et al. Durability of voretigene neparvovec for biallelic RPE65-mediated inherited retinal disease: Phase 3 results at 3 and 4 years. Ophthalmology 2021;128:10:1460-1468.
9. Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, Marshall KA et al. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: Results of Phase 1 and 3 trials. Ophthalmology 2019;126:9:1273-1285.
10. Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, Borchert M, Sengillo JD, Mendoza C, Berrocal AM et al. Perifoveal chorioretinal atrophy after subretinal voretigene neparvovec-rzyl for RPE65-mediated Leber congenital amaurosis. Ophthalmol Retina 2022;6:1:58-64.
11. Sengillo JD, Gregori NZ, Sisk RA, Weng CY, Berrocal AM, Davis JL, Mendoza-Santiesteban CE, Zheng DD, Feuer WJ, Lam BL. Visual acuity, retinal morphology, and patients’ perceptions after voretigene neparvovec-rzyl for RPE65-associated retinal disease. Ophthalmol Retina 2022;6:4:273-283.
12. Deng C, Zhao PY, Branham K, Schlegel D, Fahim AT, Jayasundera TK, Khan N, Besirli CG. Real-world outcomes of voretigene neparvovec treatment in pediatric patients with RPE65-associated Leber congenital amaurosis. Graefes Arch Clin Exp Ophthalmol. Jan 10, 2022 (online article). doi: 10.1007/s00417-021-05508-2. (Online ahead of print)
13. Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, Roman AJ, Sumaroka A, Han IC, Hochstedler MD et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med 2019;25:2:225-228.
14. Philippidis A. Editas early data for CRISPR therapy EDIT-101 shows efficacy “signals” in two patients. https://www.genengnews.com/news/editas-early-data-for-crispr-therapy-edit-101-shows-efficacy-signals-in-two-patients/. Online article. Accessed April 11, 2022.
15. Lam BL, Davis JL, Gregori NZ: Choroideremia gene therapy. Int Ophthalmol Clin 2021;61:4:185-193.
16. Lam BL, Davis JL, Gregori NZ, MacLaren RE, Girach A, Verriotto JD, Rodriguez B, Rosa PR, Zhang X, Feuer WJ. Choroideremia gene therapy Phase 2 clinical trial: 24-Month results. Am J Ophthalmol 2019;197:65-73.
17. Dimopoulos IS, Hoang SC, Radziwon A, Binczyk NM, Seabra MC, MacLaren RE, Somani R, Tennant MTS, MacDonald IM. Two-year results after AAV2-mediated gene therapy for choroideremia: The Alberta experience. Am J Ophthalmol 2018;193:130-142.
18. Morgan JIW, Jiang YY, Vergilio GK, Serrano LW, Pearson DJ, Bennett J, Maguire AM, Aleman TS. Short-term assessment of subfoveal injection of adeno-associated virus-mediated hCHM gene augmentation in choroideremia using adaptive optics ophthalmoscopy. JAMA Ophthalmol 2022. Mar 10;e220158. doi: 10.1001/jamaophthalmol.2022.0158. (Online ahead of print)
19. Lu LJ, Liu J, Adelman RA. Novel therapeutics for Stargardt disease. Graefes Arch Clin Exp Ophthalmol 2017;255:6:1057-1062.
20. Parker MA, Erker LR, Audo I, Choi D, Mohand-Said S, Sestakauskas K, Benoit P, Appelqvist T, Krahmer M, Segaut-Prevost C et al. Three-year safety results of SAR422459 (EIAV-ABCA4) gene therapy in patients with ABCA4-associated Stargardt disease: An open-label dose-escalation phase I/IIa clinical trial, cohorts 1-5. Am J Ophthalmol 2022. Mar 3;S0002-9394(22)00069-1. doi: 10.1016/j.ajo.2022.02.013. (Online ahead of print)
21. Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 2018;66:157-186.
22. Johnson & Johnson Corporate communication. Interim six-month data of RPGR gene therapy shows significant vision improvement in patients living with X-Linked Retinitis Pigmentosa. https://www.jnj.com/interim-six-month-data-of-rpgr-gene-therapy-shows-significant-vision-improvement-in-patients-living-with-x-linked-retinitis-pigmentosa. Accessed April 11, 2022.
23. Shughoury A, Ciulla TA, Bakall B, Pennesi ME, Kiss S, Cunningham ET, Jr. Genes and gene therapy in inherited retinal disease. Int Ophthalmol Clin 2021;61:4:3-45.
24. Ou J, Vijayasarathy C, Ziccardi L, Chen S, Zeng Y, Marangoni D, Pope JG, Bush RA, Wu Z, Li W et al. Synaptic pathology and therapeutic repair in adult retinoschisis mouse by AAV-RS1 transfer. J Clin Invest 2015;125:7:2891-2903.
25. Ye GJ, Budzynski E, Sonnentag P, Miller PE, Sharma AK, Ver Hoeve JN, Howard K, Knop DR, Neuringer M, McGill T et al. Safety and biodistribution evaluation in cynomolgus macaques of rAAV2tYF-CB-hRS1, a recombinant adeno-associated virus vector expressing retinoschisin. Hum Gene Ther Clin Dev 2015;26:3:165-176.
26. Mishra A, Vijayasarathy C, Cukras CA, Wiley HE, Sen HN, Zeng Y, Wei LL, Sieving PA. Immune function in X-linked retinoschisis subjects in an AAV8-RS1 Phase I/IIa gene therapy trial. Mol Ther 2021;29:6:2030-2040.
27. Hahn LC, van Schooneveld MJ, Wesseling NL, Florijn RJ, Ten Brink JB, Lissenberg-Witte BI, Strubbe I, Meester-Smoor MA, Thiadens AA, Diederen RM et al. X-Linked retinoschisis: Novel clinical observations and genetic spectrum in 340 patients. Ophthalmology 2022;129:2:191-202.
28. Mishra A, Sieving PA. X-linked retinoschisis and gene therapy. Int Ophthalmol Clin 2021;61:4:173-184.