Regeneron Pharmaceuticals announced that the Food and Drug Administration approved Eylea (aflibercept) Injection, known in the scientific literature as VEGF Trap-Eye, for the treatment of patients with neovascular age-related macular degeneration at a recommended dose of 2 milligrams every four weeks for the first 12 weeks, followed by 2 mg every eight weeks.
The approval was based upon the results of two Phase III clinical studies. In these studies, Eylea dosed every eight weeks, following three initial monthly injections, was clinically equivalent to the standard of care, Lucentis (ranibizumab injection) dosed every four weeks, as measured by the primary endpoint of maintenance of visual acuity (less than 15 letters of vision loss on an eye chart) over 52 weeks. The most common adverse reactions (frequency of 5 percent or more) reported in patients receiving Eylea were conjunctival hemorrhage, eye pain, cataract, vitreous detachment, vitreous floaters and increased intraocular pressure. The adverse event profile was similar to that seen with ranibizumab.
“The approval of Eylea offers a much needed new treatment option for patients with wet AMD,” said Jeffrey Heier, MD, a clinical ophthalmologist and retinal specialist at Ophthalmic Consultants of Boston, assistant professor of ophthalmology at Tufts School of Medicine, and chair of the Steering Committee for the VIEW 1 trial. “Eylea offers the potential of achieving the efficacy we’ve come to expect from current anti-VEGF agents, but with less frequent injections and no monitoring requirements. This may reduce the need for costly and time-consuming monthly office visits for patients and their caregivers.”
“This approval is an important step forward for Regeneron and for patients suffering with wet AMD, the most common cause of blindness in the U.S. in older adults,” said Leonard S. Schleifer, MD, PhD, president and CEO of Regeneron. “We thank the patients and clinical investigators who participated in our clinical studies, the FDA, and the Regeneron employees who helped make this day possible. Now that Eylea is approved, we plan to make Eylea available to patients within the next few days.”
Eylea is a recombinant fusion protein, consisting of portions of human VEGF receptors 1 and 2 extracellular domains fused to the Fc portion of human IgG1 and formulated as an iso-osmotic solution for intravitreal administration. Eylea acts as a soluble decoy receptor that binds VEGF-A and placental growth factor (PlGF) and thereby can inhibit the binding and activation of these cognate VEGF receptors.
The recommended dose for Eylea is 2 mg administered by intravitreal injection every four weeks for the first 12 weeks, followed by 2 mg once every eight weeks. Although Eylea may be dosed as frequently as 2 mg every four weeks (monthly), additional efficacy was not demonstrated when Eylea was dosed every four weeks compared to every eight weeks.
There is a potential risk of arterial thromboembolic events (ATEs) following use of intravitreal VEGF inhibitors, including Eylea, defined as nonfatal stroke, nonfatal myocardial infarction, or vascular death (including deaths of unknown cause). The incidence of ATEs with Eylea in clinical trials was low (1.8 percent).
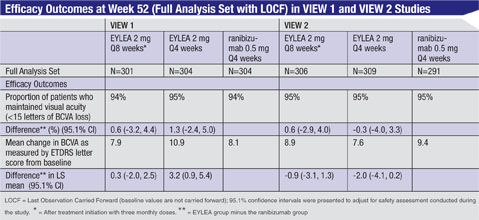 |
The safety and efficacy of Eylea were assessed in two randomized, multicenter, double-masked, active-controlled studies in patients with wet AMD. A total of 2,412 patients were treated and evaluable for efficacy (1,817 with Eylea) in the two studies (VIEW 1 and VIEW 2). In each study, patients were randomly assigned in a 1:1:1:1 ratio to one of four dosing regimens: 1) Eylea administered 2 mg every eight weeks following three initial monthly doses (Eylea 2Q8); 2) Eylea administered 2 mg every four weeks (Eylea 2Q4); 3) Eylea 0.5 mg administered every four weeks (Eylea 0.5Q4); and 4) ranibizumab administered 0.5 mg every four weeks (ranibizumab 0.5Q4). Patient ages ranged from 49 to 99 years with a mean of 76 years.
In both studies, the primary efficacy endpoint was the proportion of patients who maintained vision, defined as losing fewer than 15 letters of visual acuity at week 52 compared to baseline. Data are available through week 52. Both the Eylea 2Q8 and 2Q4 dosing groups were shown to have efficacy that was clinically equivalent to the ranibizumab 0.5Q4 group for the primary endpoint.
Stimulation Therapy May Restore Vision
It has long been thought that blindness after brain lesions is irreversible and that damage to the optic nerves leads to permanent impairments in everyday activities such as reading, driving and spatial orientation.
A new study published in Brain Stimulation suggests that treating such patients with low levels of non-invasive, repetitive, transorbital alternating current stimulation (rtACS) for 10 days (30 to 40 minutes per day) significantly reduces visual impairment and markedly improves vision-related quality of life.
The results of this study showed that treatment with rtACS resulted in an average of 41 percent shrinkage of the visual field loss. The rtACS-treated patients show significantly improved visual field sizes, which was not seen in patients who received sham treatment. Actively treated patients confirmed that their “general vision” was improved. In the sham-group, visual fields and estimates of subjective visual functioning remained largely unchanged.
“Our findings are important because they show that partial blindness can be reversed. We show for the first time that partial blindness can be reduced by a short-lasting therapeutic procedure using non-invasive electrical current stimulation,” said Bernhard A. Sabel, PhD, researcher and senior author of the study
A group of 42 patients with visual impairments following optic nerve damage participated in the study. Patients were randomly assigned to either a control condition with sham stimulation or rtACS given with an alternating current stimulation device with electrodes positioned near the eyes. The length of the daily treatment session (both rtACS and sham-treatment) varied between 10 and 20 minutes for each eye, i.e., maximum 40 minutes. The optic nerve lesions were treated long after the early recovery phase (mean lesion age 5.5 years). Both patients and the experimenter evaluating the vision parameters were masked to treatment. The study’s idea was to enhance visual system plasticity by increasing synaptic strength of residual cells in the partially damaged visual system and thus improve any residual visual capacity. The study documents a considerable activation potential of residual vision following optic nerve damage. Current pulses administered to the eye in a non-invasive manner might be able to unveil this plasticity.
The researchers were particularly interested to learn if rtACS has an effect on self-estimated visual and health-related functioning as assessed by quality of life questionnaires (e.g., the National Eye Institute Visual Function Questionnaire). Vision parameters and patient reported outcomes were collected before and after the 10-day treatment course.
The findings of this study are not only of interest to basic scientists, showing that the adult visual system is more modifiable than was previously thought, but may also help develop new therapies for patients with visual field loss. Improving vision in a subjectively meaningful way is a clinical achievement that reduces the suffering of the partially blind.
Additional studies are now under way to document in patients with visual dysfunction the neurobiological basis of the rtACS effects. Furthermore, a clinical trial with larger patient groups is currently underway to replicate these findings. Finally, the use of rtACS for the treatment of hemianopia after stroke is now being explored as well.
Potential PVR Treatment
A new study conducted by investigators at the
Schepens Eye Research Institute, the Department of Ophthalmology at Harvard Medical School, and the
Massachusetts Eye and Ear Infirmary in Boston suggests that a cocktail containing reagents to neutralize a relatively small subset of vitreal growth factors and cytokines may be an effective treatment for proliferative vitreoretinopathy.
The study was published in the December issue of the American Journal of Pathology.
“We found that a combination of seven classes of growth factors and cytokines was essential for PVR to develop in an animal model of the disease,” said lead investigator
Andrius Kazlauskas, PhD, of The Schepens Eye Research Institute and the Department of Ophthalmology, Harvard Medical School. “By neutralizing them, we prevented PVR-relevant signaling and inhibited contraction of collagen gels containing primary retinal pigment epithelial cells derived from a human PVR membrane (RPEMs). These findings suggest a potential therapeutic approach to reduce the incidence of PVR in patients undergoing surgery to repair a detached retina.”
In animal models, platelet-derived growth factor receptor α (PDGFRα) is associated with PVR and strongly promotes experimental PVR. Vitreal growth factors outside of the PDGF family promote an indirect route to activate PDGFRα. Importantly, indirectly activated PDGFRα engages a characteristic set of signaling events and cellular responses that are tightly associated with PVR. This study sought to identify the factors that would induce those events, and develop therapeutic approaches to prevent patients from developing PVR.
Vitreous was obtained from normal rabbits or those in which PVR was either developing or stabilized. Normal vitreous was found to contain substantial levels of growth factors and cytokines, which change quantitatively or qualitatively as PVR develops. A set of nine growth agents was found to be most abundant and therefore most likely to contribute to PVR. Neutralizing a subset of these factors in rabbit vitreous eliminated their ability to induce PVR-relevant signaling and cellular responses. A single dose of neutralizing reagents effectively protected rabbits from developing retinal detachment. “Our in vitro approach to characterize and neutralize vitreal bioactivity accurately predicted the efficacy of an in vivo therapy,” said Dr. Kazlauskas.
To identify growth factors likely to be driving PVR in humans, the investigators quantified the level of growth factors and cytokines from human donors that had either PVR or a non-PVR retinal condition. Fourteen of the 24 agents quantified were present in large concentrations in PVR vitreous. Neutralizing just seven of these prevented vitreous-induced activation of PDGFRα. Furthermore, the cocktail also suppressed the contraction of these cells in collagen. Therefore, the same neutralization strategy that prevented PVR in rabbits also prevented human PVR vitreous from inducing PVR-relevant responses. These results strongly suggest that a dose of neutralizing reagents may also protect humans from PVR.


