Last month, the Food and Drug Administration approved Macugen (pegaptanib sodium injection) for the treatment of neovascular age-related macular degeneration. Approval of Macugen, developed by Eyetech Pharmaceuticals Inc. and Pfizer Inc., follows a priority review under the FDA's rolling submission-Pilot 1 program based on data from the companies' Phase II/III pivotal clinical trials.
Macugen is the first in a new class of ophthalmic drugs to specifically target vascular endothelial growth factor (VEGF), a protein that acts as a signal in triggering the abnormal blood vessel growth and leakage in neovascular AMD.
"Macugen is a revolutionary, breakthrough treatment for neovascular age-related macular degeneration as it targets the pathologic processes underlying the disease," said Donald J. D'Amico, MD, professor of ophthalmology at Harvard Medical School. "Preserving vision will make a significant difference to AMD patients, since AMD can severely compromise a patient's ability to function independently."
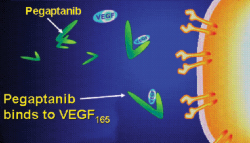 |
| Macugen (pegaptanib) selectively targets the pathogenic isoform VEGF-165. |
"The FDA's approval of Macugen for all neovascular age-related macular degeneration represents an important paradigm shift in the treatment of this devastating disease," said David R. Guyer, MD, chief executive officer and co-founder of Eyetech. "Macugen is a novel treatment based on elegant science that for the first time targets the underlying cause of the disease, which has lead to our broad AMD label, including all subtypes and sizes," Macugen is a pegylated anti-VEGF aptamer, a single strand of nucleic acid that binds with specificity to a particular target. Macugen specifically binds to VEGF 165, a protein that plays a critical role in angiogenesis (the formation of new blood vessels) and increased permeability (leakage from blood vessels), two of the primary pathological processes responsible for the vision loss associated with neovascular AMD.
Macugen is administered in a 0.3- mg dose once every six weeks by intravitreal injection. Eyetech and Pfizer plan to make the treatment available in the first quarter of 2005.
The FDA approval was based on findings from two pivotal Phase II/III randomized, multicenter, double-masked clinical trials involving approximately 1,200 patients with all subtypes of neovascular AMD. The primary efficacy endpoint was the proportion of patients protected from three line loss of visual acuity on the eye chart by week 54. Results showed that among patients receiving 0.3 mg of Macugen, 70 percent lost less than three lines of vision on the eye chart, compared with 55 percent of patients receiving control treatment (p< .0001). The results demonstrated a 27 percent relative treatment effect for Macugen-treated patients compared to controls with respect to three-line loss. Macugen also helped limit progression to legal blindness, by 50 percent compared to controls, in the study eye. Two-year clinical data from the studies demonstrated a continued treatment benefit with Macugen.
Overall, Macugen was well-tolerated and the patients who entered the second year on the same therapy received over 90 percent of possible treatments over the two years of the study, indicating strong compliance and acceptance to therapy, according to the companies. Most of the adverse events reported over the two years were mild in severity, transient and attributed by investigators to the injection procedure rather than the study drug. For full prescribing information about Macugen, visit
macugen.com.
Visx Approved for Custom Hyperopia and Astigmatism
Visx has received FDA approval to treat hyperopia and hyperopic astigmatism with the Visx CustomVue Hyperopic LASIK procedure.
The FDA approval allows for WaveScan diagnosis and Custom Vue treatment of patients with farsightedness and astigmatism. This approval is specifically for wavefront-guided LASIK for correction of hyperopia with or without astigmatism up to +3 D MRSE, with cylinder up to +2 D.
The CustomVue procedure is the first U.S.-approved, wavefront-guided laser treatment for hyperopia. Colman Kraff, MD, principal investigator with the Visx multi-center clinical study, stated, "Visx's new CustomVue Hyperopia procedure is a significant step forward in the treatment of farsightedness. The overall quality of vision with this new procedure is so superior that I plan to treat all of my qualified patients with CustomVue Hyperopia."
A six-month evaluation of clinical study participants showed that more than four times as many people were very satisfied with their night vision after the Visx CustomVue hyperopia procedure, compared to their night vision before with glasses or contacts. The Visx clinical study results also exceeded all of the FDA required parameters for safe and effective laser vision correction.
In order to perform Visx CustomVue hyperopic LASIK procedures, physicians must complete the CustomVue Hyperopic Certification Course.
FDA Accepts B&L's NDAs for Zylet Combo, Retisert
The FDA has accepted for review the New Drug Applications for Bausch & Lomb's Retisert intravitreal implant for non-infectious uveitis affecting the posterior segment, and for the combination product, Zylet (loteprednol etabonate 0.5% and tobramycin 0.3% ophthalmic suspension).
Retisert is designed to deliver a sustained release of the well-known steroid fluocinolone acetonide for up to three years. The application has Fast Track FDA status, designed to allow for quicker, priority review of novel therapies for serious diseases for which there is an unmet medical need. The implant also has received FDA Orphan Drug designation for this indication.
Bausch & Lomb's application was selected to participate in the FDA's Continuous Marketing Application Pilot 1 Program, intended to speed review by permitting reviewable units of the application to be submitted individually on a progressive basis and obtaining early feedback from FDA on each unit. The application was completed in October. B&L expects to receive FDA notification of the application status by spring 2005 and is targeting commercialization of the product in 2005.
Also approved was the NDA for Zylet, which is indicated for the treatment of steroid-responsive inflammatory ocular conditions for which a corticosteroid is indicated and where superficial bacterial ocular infection or a risk of infection exists. The prescription eye drop will be available in the United States early in the first quarter of 2005 in three sizes, 2.5 mL, 5 mL and 10 mL.
Visiogen Accommodating Lens Moves Forward
Also in early December, the FDA approved the Investigational Device Exemption for Visiogen Inc.'s Synchrony dual optic, accommodating IOL. Visiogen will begin U.S. clinical trials with the lens during 2005. Visiogen has also developed a pre-loaded injector for delivery of the lens. "The pre-loaded injector allows the surgeon to implant the Synchrony through a small incision," said Luis Vargas, MD, medical director for Visiogen. The injector has been successfully tested in vitro and in cadaver eyes. Visiogen will introduce the injector in clinical trials outside the United States during 2005.
Alcon Submits Retaane Regulatory Applications
Alcon has submitted the third and final reviewable unit of its New Drug Application for Retaane 15 mg (anecortave acetate for depot suspension) to the FDA. The application is subject to formal acceptance by the FDA, which could take up to 45 days from the date of submission. The company also submitted its European Marketing Authorisation Application for Retaane suspension. Alcon is seeking approval of the drug as a treatment for patients with subfoveal choroidal neovascularization due to AMD.
In the United States, Retaane depot is being reviewed under the FDA's new Pilot 1 Continuous Marketing Application program for fast track designated products, which allows designated NDAs to be submitted in specified reviewable units as each is completed, with each one assigned its own six-month review target.


