Inherited retinal diseases, including retinitis pigmentosa, have been the subject of therapeutic clinical trials, including the safe delivery of gene therapy to the subretinal space, with encouraging results. Affecting approximately 14,000 individuals worldwide, RP represents one of the leading causes of vision loss, with a broad spectrum of genetic and phenotypic heterogeneity. According to RetNet (https://sph.uth.edu/retnet/), a publicly available database of genes and loci mapped to inherited retinal diseases, at least 150 genes have been associated with syndromic and non-syndromic retinitis pigmentosa.
Here, we’ll discuss the clinical findings, diagnostic modalities and current clinical trials associated with RP.
Mechanism and Findings
RP is characterized by progressive vision loss due to abnormalities of the retinal photoreceptor cells or the retinal pigment epithelial cells. Usually the disease begins with damage and loss of rod cells, leading to nyctalopia and defective dark adaptation, which are the common initial concerns for patients.1 Peripheral vision loss secondary to rod dysfunction occurs early in the disease course, although it may not be recognized by an affected individual. Secondary changes in the retinal cellular milieu due to loss of rod cells eventually result in loss of cone cells. Thus, the visual field is progressively reduced in concentric rings, leading to tunnel vision and ultimately, central field loss in end-stage disease.2
Other findings of RP include posterior subcapsular cataracts (50 percent of cases)3 and photopsias. Less-common clinical findings in patients with RP include cystoid macular edema, macular holes and epiretinal membrane formation.4 Due to the large number of causative genes and known phenotypic variance associated with RP, the disease’s clinical findings, onset and progression may differ considerably between affected individuals.
RP can be inherited in the three major mendelian patterns, but can also present in isolated or simplex cases. Classically, X-linked RP patients have an earlier onset and worse prognosis, followed by autosomal recessive patients, with intermediate outcomes; and finally, dominant forms have more variable presentations and better prognoses.
 |
The differential diagnosis for RP can be further refined by age of onset. Although most individuals are diagnosed in early adulthood,5 a distinct subset of cases falls within the early-onset spectrum of diagnoses, which include Leber congenital amaurosis and early-onset severe retinal dystrophy (EOSRD)/severe early-childhood-onset retinal dystrophy (SECORD). LCA represents the most severe phenotype, characterized by profound vision impairment at birth or during the first months of life. Because pigmentary changes may only present later in life, symptoms such as nystagmus, poor object tracking and poor pupillary responses aid in the diagnosis. Electroretinogram signals are either extinguished or severely reduced. In contrast, EOSRD /SECORD are distinguished by residual, and sometimes improving, visual acuity and function; slightly preserved ERG signals; and later onset of symptoms, generally around or before the age of 5 years.6
RP can be classified as non-syndromic (affecting only the retina) or syndromic (affecting other tissues and organs). Systemic findings often associated with syndromic RP diagnoses are summarized in Table 1. The most common form of syndromic RP is autosomal recessive Usher syndrome, the leading cause of genetic deafness-blindness. There are three types of Usher syndrome (I, II, III) with several subtypes, reflecting a combination of clinical and genetic findings. The second most common form of syndromic RP is Bardet Biedl syndrome, an autosomal recessive disorder characterized by obesity, postaxial polydactyly, cognitive impairment, hypogonadotropic hypogonadism, genitourinary defects and renal disease.
Imaging and Testing
When working up a patient, the following imaging methods and tests can help clinch the diagnosis of RP:
• Fundoscopic examination. The classic triad in RP consists of peripheral bone spicule pigment deposition, blood vessel attenuation (sometimes sclerotic vessels) and optic disc pallor. Bone spicule pigmentation usually develops in the mid-stages of the disease, starting at the mid-periphery, and moves toward the macula as the disease progresses.7 Pigmentation consists of retinal pigment epithelium cells that have detached and migrated to the inner retina after photoreceptor death.8 The attenuated vessels seen in patients with RP are believed to be due to either reduced metabolic demand or vasoconstriction and reduced blood flow resulting from a hyperoxic state after the loss of oxygen-consuming photoreceptors.9,10 Loss of the retinal nerve fiber layer occurs in the late stages of the disease; therefore, optic nerve pallor isn’t necessarily synonymous with ON atrophy. Potential causes for nerve pallor include ischemia of surrounding blood vessels due to location within a watershed area, which leads to diminishment of traditional color of the ON, or astrocytic and cotton-wool-spot-like gliosis caused by the retinal degenerative process.11
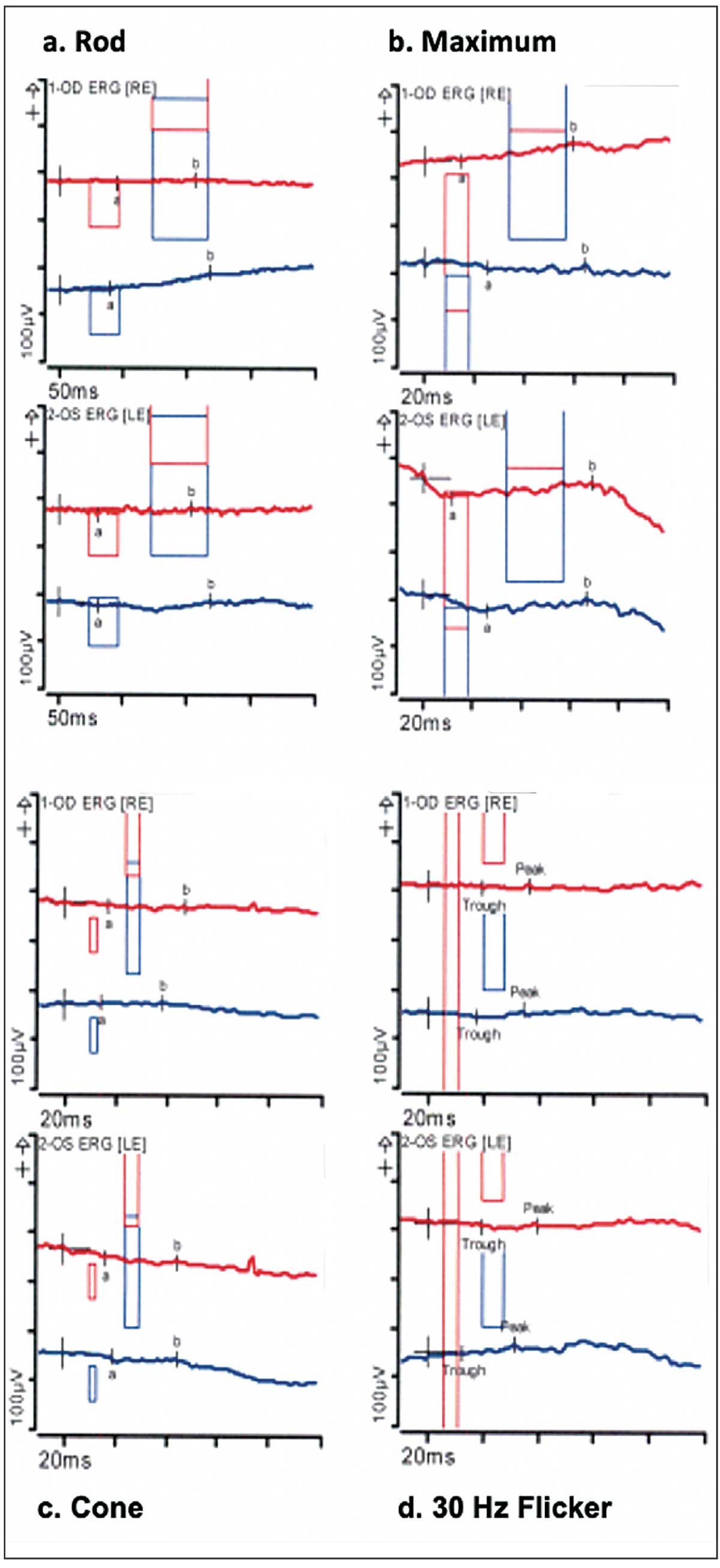 |
| Full-field ERG of a patient with severe late-stage X-linked RP showing extinguished (isoelectric) (a) rod/scotopic (dim light response in dark adaptation), (b) maximum (combined rod and cone response to maximum intensity light in dark adaptation), (c) cone/photopic (bright light response in light adaptation) and (d) 30 Hz flicker responses OU. |
• Full-field electroretinography. ERG is considered the gold-standard modality for diagnosing RP, establishing baseline function and monitoring RP progression. ERG can detect photoreceptor dysfunction even when changes on clinical exam or imaging modalities are minimal. Full-field ERG findings in RP patients include decreased rod amplitude, maximum, oscillatory, cone and flicker responses. Initially, individuals with RP have a decreased scotopic response (reflecting rod dysfunction), followed by prolonged B-wave implicit times. The eventual involvement and loss of cone photoreceptors leads to reduced amplitude of the photopic, maximum and 30 Hz flicker responses. ERG responses may be wholly extinguished in advanced stages of the disease.
Visual field testing is also suitable for establishing baseline function and monitoring disease progression. In the early stages of RP, visual field measurements show variable peripheral vision loss, progressing to a ring scotoma consistent with the tunnel vision described in the late stages of the disease.7 In a Gold-mann visual field testing study, it was found that annual rates of decline in VF area for V4e, III4e, and I4e targets were 7.5, 10.7 and 12.5 percent, respectively. Mean annual VF loss was 10.3 percent for autosomal recessive, 2.7 percent for autosomal dominant and 7.2 percent for X-linked patterns of inheritance.12
• Optical coherence tomography. OCT is used to assess retinal morphological changes in patients with RP. OCT performed early in the disease course can show disorganization of the outer retinal layers.13 With disease progression, decreased thickness of the outer nuclear layer can be observed.13 Late stages of RP are characterized by the complete loss of both the outer segment and the outer nuclear layer, with the inner retinal layers remaining relatively well preserved.14 Previous studies have described a correlation between structure captured by OCT and retinal function and show that retinal thinning (particularly foveal thinning) correlates with decreasing visual acuity and visual fields.15 OCT is also used to monitor the presence and progression of CME, macular cysts and macular holes in patients with RP. Of note, CME associated with RP can be treated with topical carbonic anhydrase inhibitors with variable results. In some cases, patients that do not respond well to topical therapy may require an oral CAI.16,17
• Fundus autofluorescence. FAF is commonly used to assess disease stage and progression. In FAF, areas of hypofluorescence representing atrophy of photoreceptors have been found to correlate with visual field defects observed by Goldmann perimetry, making them adequate complementary tests.18 Several FAF patterns are considered typical in RP, such as a hyper-autofluorescent ring or an abnormal central hyper-autofluorescence.19 This hyperfluorescent ring, also named the Robson-Holder ring, delineates the border between normal and disrupted inner and outer segment junctions, supported by OCT imaging. Significant loss of photoreceptors will be found outside the ring compared to inside it.14 The size of the ring negatively correlates with the remaining visual function, measured by perimetry.20
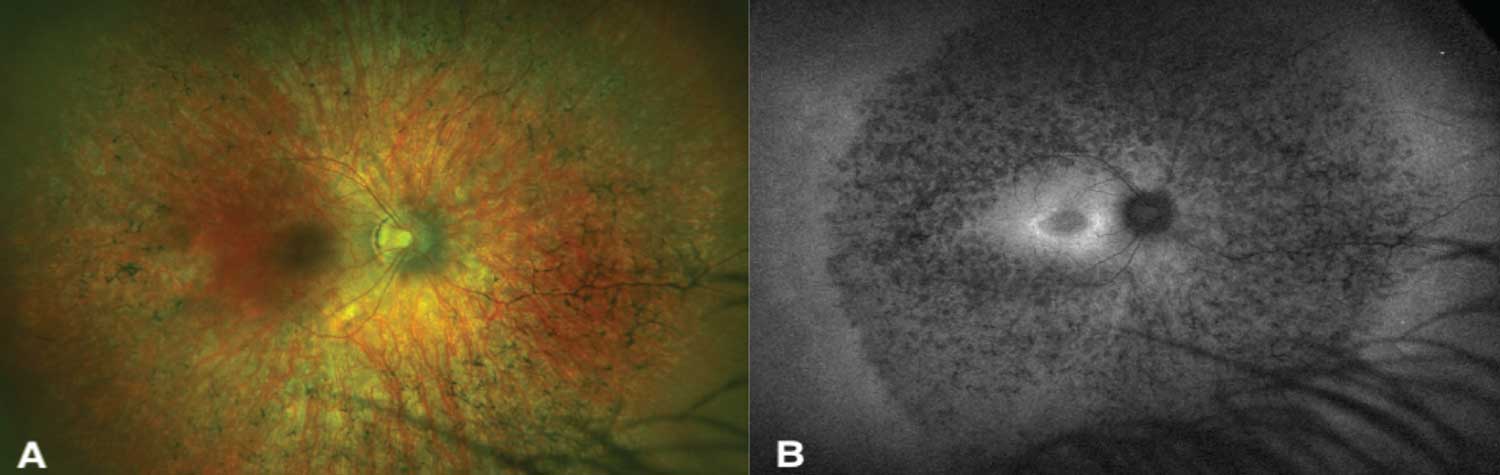 |
| Figure 1. A case of advanced RPGR-X-linked RP. A) Pseudo-color fundus photography of the right eye shows classical findings of bone spicule pigmentation, attenuated blood vessels and optic nerve pallor. B) A fundus auto-fluorescence photograph of the same eye, showing an ellipsoid-shaped hyperautofluorescent ring. |
Genetic Testing
In 2022, genetic testing is a cornerstone in the diagnosis and management of retinitis pigmentosa. In most cases, comprehensive next-generation sequencing can be performed at minimal to no cost to the patient, with multiple sponsored, open-access gene panels commercially available. Results are only available to patients and providers within weeks, providing diagnostic information that often impacts prognosis and management. However, causative genetic variants are identified in about 60 percent of cases, leaving a significant portion of cases with an undetermined genetic etiology.21 Furthermore, the ubiquitous presence of variants of uncertain significance can complicate the interpretation of results. Nevertheless, with increasing genetic testing rates and the continuous addition of genes to panels, our knowledge of the genetic etiologies of IRDs will likely continue to increase the diagnostic rates.
When considering genetic testing, ophthalmologists should be aware of some potential pitfalls such as false-positive rates, and differences in content and coverage of next-generation sequencing panels and other currently available genetic tests.22 Thus, pre-test phenotyping, consisting of obtaining a detailed medical history and reviewing diagnostic testing and imaging, is fundamental prior to ordering genetic testing in order to interpret results accurately.22 Additionally, working with a genetic counselor who can assist in panel selection and communicating testing results to patients, as well as education on prognosis, inheritance and natural history, should be considered and is highly recommended when available.
Vitamin Supplementation
Several studies have evaluated the use of vitamin supplementation, particularly vitamin A, in RP patients. Although early trials reported clinical benefits, later studies have shown that the efficacy of vitamin A in slowing disease progression is inconclusive.23 Furthermore, some cases of suspected RP can present similarly to Stargardt disease, which is caused by mutations in the ABCA4 gene that result in vitamin A metabolism dysfunction, leading to the accumulation of the A2E by-product and lipofuscin deposits in the macula.24 Thus, until this condition has been ruled out, particularly by genetic testing, patients should refrain from using any vitamin A or vitamin A-containing food, to decrease the risk of worsening. Patients can be advised to use supplements such as lutein and zeaxanthin (found in AREDS2 formulations such as PreserVision and Ocuvite) commonly offered for macular degeneration, which have been found to aid in slowing disease progression and are safe for patients with Stargardt disease.
Luxturna Arrives
In 2017, Luxturna (voretigene neparvovec, Spark Therapeutics), an adeno-associated virus (AAV2) vector carrying an RPE65 cDNA, became the first FDA-approved gene therapy for an IRD. Designed for the treatment of RPE65-related Leber’s Congenital Amaurosis and a small percentage of cases of autosomal recessive RP (~2 percent),25 the pivotal Phase III clinical trial revealed that the medication met its primary endpoint, improving multi-illuminance mobility testing with adequate efficacy and safety profile after one year.
Clinical Trials
Following the significant milestone represented by Luxturna, clinical trials evaluating similar interventions for other genotypes of RP are being developed or are currently underway. Current trials use gene augmentation, silencing, editing, optogenetics and stem cell replacement approaches. Here, we will briefly highlight several completed and ongoing clinical trials evaluating new therapies for RP (See Table 2 below).
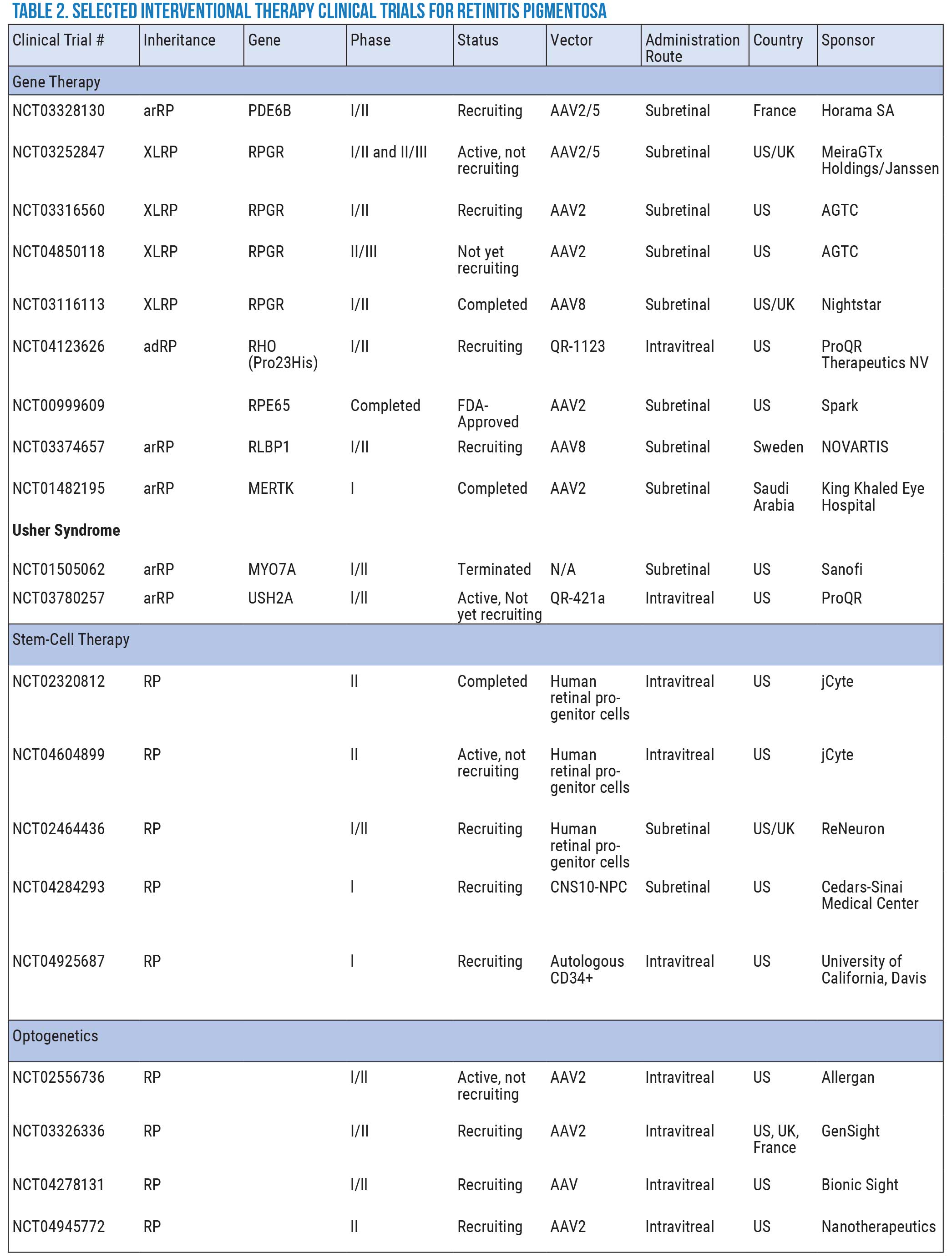 Click image to enlarge. |
• Gene therapy. Genetic therapy remains the most promising approach for the treatment of IRDs. Therapies can be delivered to the subretinal space through viral vectors, commonly AAVs, to provide a functional copy of a gene (augmentation) or to correct a mutation (editing). The retina’s immune-privileged environment, as well as its accessibility for surgical procedures, make it a suitable candidate for this type of approach.
Clinical trials assessing therapies that target various specific genes implicated in RP are currently under way:
— RHO-related disease due to P23H mutations, QR-1123 (AURORA trial, ProQR Therapeutics NV). QR-1123 is an antisense oligonucleotide (ASO) designed to exclusively target the mutant P23H mRNA in RHO, which has been reported to account for 10 percent of autosomal dominant cases in the United States.26 ASOs have been shown to reduce the expression of the mutant mRNA through specific nucleotide pairings using a mechanism called RNase H mediated cleavage, while preserving the expression of the wild type.27 In vivo mouse models showed a 40-percent reduction of P23H mRNA with preservation of WT RHO and improvement in scotopic responses in ONL thickness.28 The AURORA trial is currently recruiting patients 18 years of age or older to evaluate the safety and tolerability of intravitreal injections of QR-1123, through open-label single-dose cohorts and randomized repeat-dose cohorts.29
— USH2A-related disease, QR-421a (STELLAR trial, ProQR Therapeutics NV). Similarly, QR-421a is an RNA-based therapy designed to treat Usher syndrome type II. This therapy uses an exon-skipping approach where ASOs bind specifically to the USH2A RNA and exclude exon 13 from the RNA, leading to retinal cells that produce a shorter but functional copy of the USH2A protein. Interim results from their STELLAR trial showed that the therapy was adequately tolerated across all doses with no significant adverse events observed. Mean visual acuity improved by six letters in treated patients, while patients with advanced disease improved by 9.3 letters. At week 12, the mean change from baseline in the number of loci with improved static perimetry was 9.2 loci in treated eyes versus 6.1 in untreated eyes. OCT imaging also demonstrated stabilization of retinal architecture in the treated eyes versus sham and untreated eyes. (Results available at https://www.proqr.com/files/2021-09/Euretina%20%282021%29%20DG%20Birch.%20QR-421a%20Stellar%20results.pdf)
— RPGR-related disease, MGT009 (Mereira/Janssen). MGT009 is a recombinant AAV2/5 vector designed to deliver functional copies of the open reading frame 15 (ORF15) of the RPGR gene to the subretinal space in males with XLRP. A Phase I/II trial is currently enrolling 46 patients, five years of age or older, to study the dose-escalated safety and efficacy of the drug with an 18-month timeframe.30 Initial 12-month results showed that four patients in the intermediate-dose cohorts achieved clinically meaningful improvements in retinal sensitivity (1.05 dB; [90% CI: 0.81, 1.29]) and visual field progression rate (1.26 dB-sr/year; [90% CI: 0.65, 1.86]). Patients in the low- and intermediate-dose cohorts (n=6) also achieved significant improvements in their vision-guided mobility maze evaluation at low light levels with a -16.1 seconds (90% CI: 9.91, 22.1) difference between treated and untreated eyes. There were no reports of dose-limiting events, although signs of inflammation were observed in two of three patients in the high-dose cohort, successfully managed with steroids.31 The sponsor plans to proceed with a Phase III trial after completing the 18-month Phase I/II trial.
— RPGR-related disease, AGTC-501 (AGTC). AGTC-501 is also a recombinant AAV2 vector, administered by subretinal injection in affected RPGR XLRP patients with mutations within exons 1-14 or ORF15. Thirty participants, 6 to 50 years old, were enrolled in the dose-escalation portion of the Phase l/ll trial. Participation included a total of 15 visits over approximately 36 months and long-term follow-up evaluations annually at years four and five.32 Preliminary results were presented at the 2021 AAO meeting: AGTC-501 was well-tolerated across a wide dose range, with minimal adverse effects such as ocular inflammation and blurred vision. Treatment with AGTC-501 resulted in a statistically significant improvement in best-corrected visual acuity (BCVA) of ≥5 letters (9 of 13 subjects; p≤0.005). Patients were considered responders if an improvement of at least 7 decibels (dB) in at least five loci within the central 36 loci macular area (p≤0.05) was obtained by macular integrity assessment (MAIA) microperimetry. At 12 months, four out of eight patients were considered responders.33 When the Phase I/II phase is completed, a planned Phase II/III will randomize 63 participants in order to compare two doses (low and high) of AGTC-501 and an untreated control group over a 12-month time frame.
• Optogenetics. This is an emerging technology that employs optimized opsins, light-sensitive proteins that can modulate neural activity in retinal cells, using existing neural synapses to act as artificial photoreceptors.34
Opsin genes, such as channelrhodopsin, can be transfected into non-photoreceptor cells, such as retinal ganglion cells, through commonly used vectors, such as AAV, via subretinal injection. Four companies (Allergan, Gensight, Nanoscope Therapeutics and Bionic Sight) are actively recruiting patients for Phase I/II clinical trials to study their respective vectors’ safety and efficacy profiles. Gensight’s trial will test the combined intervention of their GS030A vector, injected into the worse-seeing eye, and a pair of specialized light-stimulating glasses (GS030-Medical Device) tasked with amplifying external visual stimuli to the transfected retina.35
Recently, Nanoscope Therapeutics shared results for its Phase I trial evaluating the safety and efficacy of MCO-010, an AAV2 vector containing multi-characteristic opsin (MCO) that doesn’t need a combination of implants or goggles, in patients with advanced disease. The results showed that six out of seven (86 percent) high-dose (3.5 × 1011 VG/eye) MCO-therapy subjects gained >0.3 logMAR (15 letters) at 52 weeks.36 The sponsor is currently recruiting patients for its randomized, double-masked Phase II RESTORE trial to assess the efficacy and safety profiles of the therapy with a timeframe of one year.37
Trials for optogenetics are all still in the early stages, with reports for only a limited number of treated patients available. Thus, future studies will help substantiate their safety and efficacy in treating RP.
• Stem cell therapy. Several types of stem cells, such as retinal progenitor, embryonic, induced pluripotent and mesenchymal, are being studied as a potential treatment modality in retinal dystrophies. Pre-clinical studies have shown advantageous effects of stem cell treatment, such as replacing damaged cells, adding nutritional support to remaining functioning cells, protecting retinal vascularity and promoting synaptic connections.38 It’s important to consider that each type of stem cell has a unique set of advantages and disadvantages.
Multiple clinical trials evaluating the safety and efficacy of stem cell therapy from various countries have had varying results.38 Currently, several clinical trials in the United States, sponsored by companies and institutions such as jCyte, ReNeuron, Cedars Sinai Medical Center and UC Davis, are holding trials to assess the use of human retinal progenitor, neural progenitor and mesenchymal stem cells, respectively, for the treatment of RP.
One sponsor, jCyte, reported the results of its Phase IIb randomized trial evaluating the efficacy of intravitreal administration of a 2-milion to 6-million-cell dose of its allogeneic human retinal progenitor cell (hRPCs) therapy, jCell, in 74 patients with RP.39 Results presented at the AAO 2021 meeting showed that patients with a central VF diameter >20 degrees had a significant improvement in visual function as measured by the primary endpoint (BCVA), which changed from baseline by 15.6 letters above sham (p=0.029).40 Differences between jCell and sham treatment groups were also found in secondary endpoints such as contrast sensitivity (CS): +242 percent; kinetic visual field (KVF): +317 percent; and the low luminance mobility test (LLMT): +1 Critical Illumination Level. The presentation highlighted the dependence of cone photoreceptors on the rod-derived cone viability factor (RdCVF), released by rod photoreceptors, which results in patients with more surviving adjacent rod photoreceptors having a far greater potential for restoration of cone photoreceptor function. Adverse events were generally minor and transient with only one serious Grade 3 adverse event (ocular hypertension) in the 3.0×106 hRPC during the 12-month study period.40
Future Directions
Gene editing employing the CRISPR/Cas9 system is an emerging approach for future therapies for IRDs. In brief, the CRISPR system acts as molecular scissors that cut out a portion of the damaged gene and replace it with a wild-type sequence. Results of in vitro retinal cell and in vivo mouse studies are promising, showing a slowed progression of RP.41
Recently, EDITAS Medicine released initial results from its ongoing BRILLIANCE trial for the safety and efficacy parameters of their EDIT-101 therapy to treat CEP290-related Leber congenital amaurosis. After an initial 15-month period, results showed a satisfactory safety profile with no drug-related toxicities. Two out of five patients in the medium-dose cohort saw improvement in secondary endpoints such as BCVA, FST and mobility navigation.42 This method can be explored in the treatment of many other inherited retinal diseases if studies continue to produce favorable safety and efficacy results.
In conclusion, retinitis pigmentosa remains a leading cause of hereditary visual impairment. Diagnosis is made through a combination of clinical symptoms, diagnostic and functional assessments, and genetic testing. While treatments remain elusive, extensive research, including late-phase therapeutic clinical trials involving genetic and stem cell approaches, is under way. Initial results are promising, and we anticipate significant progress will be made in the coming years.
Dr. Regillo is the director of the Retina Service of Wills Eye Hospital, a professor of ophthalmology at Thomas Jefferson University School of Medicine and the principle investigator for numerous major international clinical trials.
Dr. Yonekawa is an assistant professor of ophthalmology at Sidney Kimmel Medical College at Thomas Jefferson University. He serves on the Education Committee of the American Society of Retina Specialists and on the Executive Committee for the Vit Buckle Society, where he is also the vice president for academic programming.
1. Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. GeneReviews. Published online January 19, 2017. Accessed December 21, 2021. https://www.ncbi.nlm.nih.gov/books/NBK1417/
2. Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 2018;66:157-186.
3. Fishman GA, Anderson RJ, Lourenco P. Prevalence of posterior subcapsular lens opacities in patients with retinitis pigmentosa. Br J Ophthalmol 1985;69:4:263-266.
4. Hagiwara A, Yamamoto S, Ogata K, et al. Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol 2011;89:2.
5. Tsujikawa M, Wada Y, Sukegawa M, et al. Age at onset curves of retinitis pigmentosa. Arch Ophthalmol 2008;126:3:337-340.
6. Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol 2017;101:9:1147-1154.
7. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis 2006;1:1.
8. Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res 1998;17:2:175-205.
9. Grunwald JE, Maguire AM, Dupont J. Retinal hemodynamics in retinitis pigmentosa. Am J Ophthalmol 1996;122:4:502-508.
10. Yu DY, Cringle SJ. Retinal degeneration and local oxygen metabolism. Exp Eye Res 2005;80:6:745-751.
11. Ilhan C, Citirik M. Glial proliferation and atrophy: Two poles of optic disc in patients with retinitis pigmentosa. J Curr Ophthalmol 2019;31:4:416-421.
12. Xu M, Zhai Y, MacDonald IM. Visual field progression in retinitis pigmentosa. Investig Ophthalmol Vis Sci 2020;61:6.
13. Liu G, Liu X, Li H, Du Q, Wang F. Optical coherence tomographic analysis of retina in retinitis pigmentosa patients. Ophthalmic Res 2016;56:3:111-122. doi:10.1159/000445063
14. Hood DC, Lazow MA, Locke KG, Greenstein VC, Birch DG. The transition zone between healthy and diseased retina in patients with retinitis pigmentosa. Investig Ophthalmol Vis Sci 2011;52:1:101-108.
15. Battu R, Khanna A, Hegde B, Berendschot TTJM, Grover S, Schouten JSAG. Correlation of structure and function of the macula in patients with retinitis pigmentosa. Eye 2015;29:7:895-901.
16. Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and Usher syndrome. Arch Ophthalmol 2010;128:9:1146-1150.
17. Huang Q, Chen R, Lin X, Xiang Z. Efficacy of carbonic anhydrase inhibitors in management of cystoid macular edema in retinitis pigmentosa: A meta-analysis. PLoS One 2017;12:10.
18. Ogura S, Yasukawa T, Kato A, et al. Wide-field fundus autofluorescence imaging to evaluate retinal function in patients with retinitis pigmentosa. Am J Ophthalmol 2014;158:5:1093-1098.
19. Murakami T, Akimoto M, Ooto S, et al. Association between abnormal autofluorescence and photoreceptor disorganization in retinitis pigmentosa. Am J Ophthalmol 2008;145:4:687-694.
20. Popović P, Jarc-Vidmar M, Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefe’s Arch Clin Exp Ophthalmol 2005;243:10:1018-1027.
21. Huang H, Chen Y, Chen H, et al. Systematic evaluation of a targeted gene capture sequencing panel for molecular diagnosis of retinitis pigmentosa. PLoS One 2018;13:4:e0185237. doi:10.1371/JOURNAL.PONE.0185237.
22. Pulido JS, Procopio R, Davila HJ, Bello N, Ku C, Pennesi ME, Yang P, Nagiel A, Mahroo OA, Aleman TS, Salido EM, Reynolds M. IRD panels—Caveat emptor—truly know your IRD panel. Retina 2022;42:1:1-3.
23. Zhao Y, Feng K, Liu R, Pan J, Zhang L, Lu X. Vitamins and mineral supplements for retinitis pigmentosa. J Ophthalmol. 2019;2019. doi:10.1155/2019/8524607
24. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 2017;101:1:25.
25. Parmeggiani F, S. Sorrentino F, Ponzin D, Barbaro V, Ferrari S, Di Iorio E. Retinitis pigmentosa: Genes and disease mechanisms. Curr Genomics 2011;12:4:238.
26. Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, Gire AI, Seaman R, Duzkale H, Spellicy CJ, Zhu J, Shankar SP, Daiger SP. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Invest Ophthalmol Vis Sci 2006;47:7:3052.
27. Scoles DR, Minikel E V., Pulst SM. Antisense oligonucleotides: A primer. Neurol Genet 2019;5:2:323.
28. Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, Watt A, Freier S, Hung G, Adamson PS, Guo S, Monia BP, LaVail MM, McCaleb ML. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci 2015;56:11:6362-6375.
29. ProQR Therapeutics-NCT04123626. A study to evaluate the safety and tolerability of QR-1123 in subjects with autosomal dominant retinitis pigmentosa due to the P23H mutation in the RHO gene. ClinicalTrials.gov. Accessed November 24, 2021. https://www.clinicaltrials.gov/ct2/show/NCT04123626?term=gene+therapy&cond=RHO&draw=2&rank=1.
30. MeiraGTx UK II Ltd-NCT03252847. Gene therapy for X-linked retinitis pigmentosa (XLRP) retinitis pigmentosa GTPase regulator (RPGR). ClinicalTrials.gov. Accessed November 24, 2021. https://clinicaltrials.gov/ct2/show/NCT03252847?term=adeno+associated+virus&cond=Retinitis+Pigmentosa&draw=2&rank=3.
31. Johnson & Johnson. Interim six-month data of RPGR gene therapy shows significant vision improvement in patients living with X-Linked Retinitis Pigmentosa. https://www.jnj.com/interim-six-month-data-of-rpgr-gene-therapy-shows-significant-vision-improvement-in-patients-living-with-x-linked-retinitis-pigmentosa. Accessed November 25, 2021.
32. AGTC. A clinical trial evaluating the safety and efficacy of a single subretinal injection of AGTC-501 in participants with X-linked retinitis pigmentosa caused by RPGR mutations. ClinicalTrials.gov. https://www.clinicaltrials.gov/ct2/show/NCT04850118?term=agtc&draw=2&rank=1. Accessed November 25, 2021.
33. AGTC. AGTC presents data from ongoing Phase 1/2 Trial of AGTC-501. https://www.globenewswire.com/news-release/2021/11/12/2333372/30580/en/AGTC-Presents-Data-from-Ongoing-Phase-1-2-Trial-of-AGTC-501-at-Upcoming-American-Academy-of-Ophthalmology-2021-Annual-Meeting.html. Accessed November 25, 2021.
34. Duebel J, Marazova K, Sahel JA. Optogenetics. Curr Opin Ophthalmol 2015;26:3:226-232.
35. Martel J, Boulanger-Scemama E, Esposti S. Dose-escalation study to evaluate the safety and tolerability of GS030 in subjects with retinitis pigmentosa. ClinicalTrials.gov. GenSight Biologics. https://clinicaltrials.gov/ct2/show/NCT03326336. Accessed November 25, 2021.
36. Nanoscope Therapeutics. Nanoscope’s optogenetic gene therapy restores clinically meaningful vision in 11 patients blinded by retinitis pigmentosa. https://nanostherapeutics.com/2021/06/03/nanoscopes-optogenetic-gene-therapy-restores-clinically-meaningful-vision/. Accessed November 16, 2021.
37. Nanoscope Therapeutics. Efficacy and safety of vmco-010 optogenetic therapy in adults with retinitis pigmentosa [RESTORE]. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04945772?term=optogenetics&draw=2&rank=1. Accessed November 25, 2021.
38. Wang Y, Tang Z, Gu P. Stem/progenitor cell-based transplantation for retinal degeneration: A review of clinical trials. Cell Death Dis 2020;11:9:1-14.
39. jCyte. Safety and efficacy of intravitreal injection of human retinal progenitor cells in adults with retinitis pigmentosa. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03073733. Accessed November 25, 2021.
40. jCyte. jCyte identifies key anatomical biomarker predictive of substantial restoration of visual function in retinitis pigmentosa patients treated with jCell therapy. https://www.businesswire.com/news/home/20211115005800/en/jCyte-Identifies-Key-Anatomical-Biomarker-Predictive-of-Substantial-Restoration-of-Visual-Function-in-Retinitis-Pigmentosa-Patients-Treated-With-jCell-Therapy. Accessed November 16, 2021.
41. Gumerson JD, Alsufyani A, Yu W, Lei J, Sun X, DongL, Wu Z, Li T. Restoration of RPGR expression in vivo using CRISPR/Cas9 gene editing. Gene Ther. Published online July 14, 2021:1-13. doi:10.1038/s41434-021-00258-6
42. Editas. Editas Medicine announces positive initial clinical data from ongoing Phase 1/2 BRILLIANCE clinical trial of EDIT-101 for LCA10. Editas Medicine. Accessed December 14, 2021. https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-positive-initial-clinical-data-ongoing.