Imitation is the sincerest form of flattery. It also may be the sincerest way to make effective medications more affordable for patients with retinal conditions in the form of biosimilars, which can offer similar efficacy as their reference products at about 40 percent of the price. Two biosimilars for ophthalmic indications have been approved in the United States already and more appear to be on the way. Here, we’ll take a look at the development of biosimilars in retina and the safety and efficacy of agents both approved and in development.
Biosimilar Background
 |
| Cimerli was deemed interchangeable with ranibizumab. |
In the United States, intravitreal anti-VEGF agents approved by the Food and Drug Administration include not only ranibizumab, aflibercept, brolucizumab and faricimab, but also recently approved biosimilar anti-VEGF agents. All of these anti-VEGFs are biotechnology-derived protein products, classified as biologics.1 As such, their production entails the use of living cells with culture media and various excipients that result in slight differences between lots; these differences are accepted as normal and expected. The FDA assesses the manufacturing process and the manufacturer’s strategies to control and monitor these within-product variations in order to produce a biologic with consistent clinical performance. The patented and FDA-approved biological product is termed an FDA “reference product,” and serves as the standard to which biosimilars are compared. Examples of reference products include ranibizumab, aflibercept and bevacizumab. The expiration of the patents for ranibizumab and bevacizumab in the United States (2020 and 2019, respectively) facilitated the development of less costly biosimilars.2
A biosimilar is a biologic product that’s very similar to the reference product, with no clinically meaningful difference in terms of safety, purity or potency.3 Guidelines for the approval of biosimilars were first created in Europe, where the European Medicines Agency created an abbreviated registration process in 2005-2006, resulting in the EMA’s first approved biosimilar, a somatropin biosimilar called Omnitrope, in 2006.2 In the United States, The U.S. Patient Protection and Affordable Care Act created an abbreviated licensure pathway for biosimilars (351(k) pathway) under the Biologics Price Competition and Innovation Act of 2009 (BPCI Act), which was signed into law in 2010. The final guidance on implementation of the 351(k) pathway was in 2015.4 The FDA approved its first biosimilar in 2015.
The driving force for creation of biosimilars is the lower cost, which can lead to improved affordability and accessibility to the anti-VEGFs. Biosimilars cost less than the reference product because research and development costs are cheaper. This is primarily due to lower clinical trial costs since the FDA requires much less extensive clinical trials for biosimilar approval compared to approval of a reference product. Indeed, current biosimilars cost about 40 percent less than the reference product.2
 |
Approving a Biosimilar
The FDA approval process for a biosimilar differs from that of the reference product. In contrast to the reference product, which must demonstrate efficacy and safety in costly clinical trials, a biosimilar just needs to show it’s similar to the reference product and that its treatment outcomes wouldn’t be expected to differ from the reference product. The manufacturer must show that high similarity to the reference product’s biological activity, purity and structure. The primary endpoint is selected as a sensitivity endpoint, which shows the highest change from baseline for the treatment response. For anti-VEGFs, the most rapid change in visual acuity occurs within the first eight to 12 weeks of initiating therapy. Thus, the non-inferiority studies for anti-VEGF biosimilars use a primary endpoint of change in mean visual acuity from baseline at eight to 12 weeks.
 Byooviz was the first biosimilar FDA Byooviz was the first biosimilar FDAapproved for macular degeneration. |
Unlike non-biologically derived drugs for which generic drugs copy an identical chemical formula, biologics are produced by living cells and there isn’t a chemical formula or recipe for production. Differences in clinically inactive components are acceptable; these include differences in stabilizers and buffers. Safety is demonstrated through human pharmacokinetic (exposure) and efficacy through pharmacodynamic (response) studies. In addition, an assessment of clinical immunogenicity as compared to the reference drug is also needed as antidrug antibodies (ADA) develop in response to intravitreal injection of anti-VEGFs into the vitreous.5
In the approval process for a biosimilar, extrapolation to disease indications that weren’t studied in a clinical trial may be granted if FDA review of the data shows no differences between the biosimilar and the reference product. The FDA works with the manufacturer to determine what data are needed for extrapolation.6 Designation of “interchangeable” may also be granted by the FDA if the data shows the biosimilar would be expected to result in the same clinical result as the reference product. Usually this includes studies in which repeated doses are given and for which safety and effectiveness are shown when switching back and forth between the reference and the biosimilar product. It’s important to be aware that an interchangeable biosimilar can be substituted for the reference product by a pharmacy without the physician being aware of this substitution. These laws pertaining to pharmacy-level substitution vary from state to state.
Biosimilars’ Reception
Introduction of biosimilars into medicine has been met with some initial resistance. Uptake of the use of biosimilars is typically slow initially and then accelerates as providers become more educated about them.7 A contributing factor to this slow uptake is known as the “nocebo” effect, which is defined as the “incitement or the worsening of symptoms induced by any negative attitude from non-pharmacological therapeutic intervention, sham or active therapies.”8 In order to combat this factor, there needs to be effective, clear, data-driven communication of the biosimilar by the physician to the patient. Positive framing, data about similarity and expected outcomes, and a unified office approach to the biosimilar should help decrease the nocebo effect.
The FDA has implemented initiatives to improve access to biosimilars in the United States. The agency’s Biosimilars Action Plan (July 2018) was created to improve the efficiency of development and approval of biosimilars, including supporting education and communication to improve understanding of the drugs.3 In other areas of medicine, such as oncology, biosimilars are commonplace.
In the following sections, we’ll discuss the biosimilars in the retina space.
Ranibizumab Biosimilars
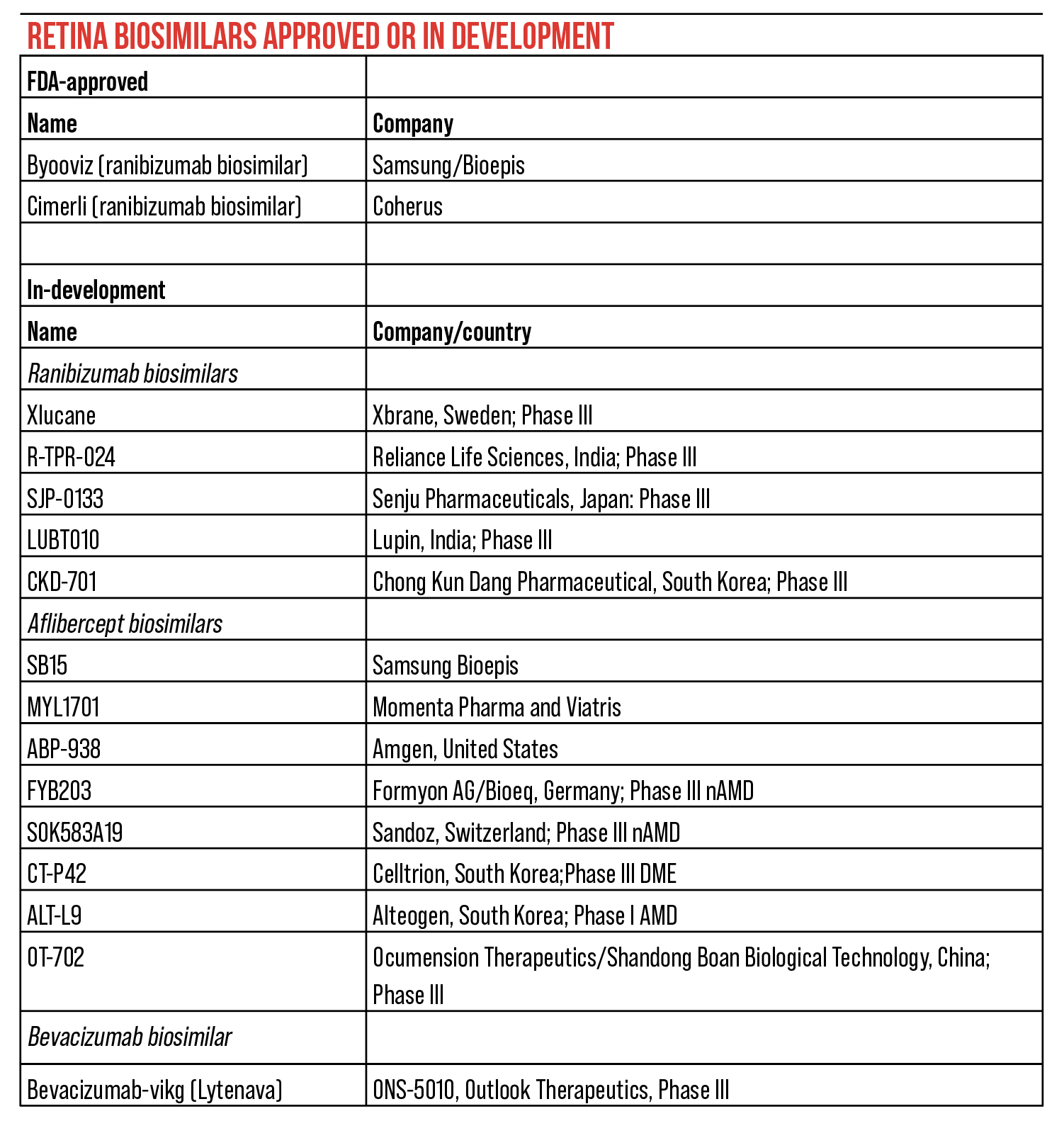
There are several ranibizumab biosimilars, some of which are approved in the United States: Byooviz (SB11) and Cimerli (FYB 201). Other biosimilars in development include Xlucane (Sweden), R-TPR-024 (India), SJP-0133 (Japan), LUBT010 (India) and CKD-701 (South Korea).9
In 2015, India became the first country to launch a biosimilar of ranibizumab, known as Razumab, (Intas Pharmaceuticals; Ahmedabad, Gujarat, India) for the treatment of neovascular AMD, diabetic macular edema and retinal vein occlusion.10-15 The Phase III clinical trial was for the treatment of AMD. Subsequent retrospective studies included RE-ENACT12-14 and CESAR (Clinical Efficacy and Safety of Razumab).15 The introduction of this biosimilar was associated with inflammatory reactions, specifically sterile endophthalmitis, from the use of specific batches of Razumab.16 These were found to be due to higher endotoxin levels in the buffer used for manufacturing. Since then, an extensive review has demonstrated good results for Razumab in India.17
The first FDA-approved biosimilar was for ranibizumab. Ranibizumab-nuna (SB11, Byooviz; Biogen/Samsung Bioepis, South Korea) was approved in September 2021 for treatment of nAMD, macular edema from RVO and myopic choroidal neovascularization. In the Phase III non-inferiority clinical trial of SB11 in nAMD, 705 patients were randomized (1:1) to receive SB11 or reference ranibizumab monthly injections (0.5 mg) in an equivalence study with a primary equivalence endpoint was at week eight and follow-up through week 24.18 Equivalence was demonstrated for safety, efficacy and immunogenicity. Extended follow-up at week 52 for 634 patients who continued to receive treatment up to week 48 continued to show equivalence for efficacy and safety. The Least Squares (LS) mean change in best-corrected visual acuity from baseline at week 52 was 9.79 letters for SB11, compared with 10.41 letters for reference ranibizumab (difference: -0.62, [90% CI: -2.092, 0.857]). The LS mean change in central subfield thickness (CST) was −139.55 µm for SB11 vs −124.46 µm for reference ranibizumab (difference: -15.09, [95% CI, -25.617, -4.563]). At all timepoints studied up to week 52, pharmacokinetic, safety and immunogenicity profiles of SB11 were comparable to reference ranibizumab.19
In October 2022, FYB-201, ranibizumab-eqrn (Cimerli, Coherus BioSciences) became FDA approved as the only interchangeable biosimilar to the reference ranibizumab (Lucentis, Genentech) for all ranibizumab indications. It’s available in both 0.3-mg and 0.5-mg dosages for nAMD, macular edema following RVO, DME, diabetic retinopathy and mCNV.20 The interchangeability designation resulted from the FDA assessment that ranibizumab-eqrn met FDA safety, efficacy and quality standards to the reference product. This included comprehensive studies involving analytical, preclinical and clinical programs to confirm equivalent safety and efficacy to ranibizumab. Cimerli was granted 12 months of interchangeability exclusivity.
In the Phase III COLUMBUS-AMD study, 477 patients were randomized to receive monthly ranibizumab-eqrn 0.5 mg or reference ranibizumab 0.5 mg in a non-inferiority design.21 The primary endpoint was change in best-corrected visual acuity after eight weeks, with an equivalence margin of three letters. Ranibizumab-eqrn met the primary endpoint with an improvement of 5.1 letters as compared with reference ranibizumab showing an improvement of 5.6 letters (-0.4 difference, 90% CI -1.6 to 0.9). Ocular and systemic safety profiles were similar between the treatment groups. Secondary endpoints included mean change in BCVA from baseline, change from baseline in retinal thickness at 48 weeks, safety and immunogenicity. The overall safety and immunogenicity profile was comparable to ranibizumab’s. Based on the totality of evidence, ranibizumab-eqrn demonstrated that clinical outcomes are expected to be the same for any given patient across all indications.
Aflibercept Biosimilars
SB15 (Samsung Bioepis) and MYL1701 (Momenta Pharmaceuticals and Viatris) are two aflibercept biosimilars in development that currently have some clinical trial results available. Other aflibercept biosimilars being developed include:
- ABP-938 (Amgen, U.S.);
- FYB203 (Formycon AG/ Bioeq, Germany; Phase III nAMD MAGELLAN study);
- SOK583A19 (Sandoz, Switzerland; Phase III nAMD MYLIGHT study);
- CT-P42 (Celltrion, South Korea; Phase III DME);
- ALT-L9 (Alteogen, South Korea; Phase I nAMD); and
- OT-702 (Ocumension Therapeutics/Shandong Boan Biological Technology, China; Phase III).9
SB15 is being studied in a Phase III clinical trial in treatment-naïve nAMD patients. Patients were randomized to receive either SB15 or reference product aflibercept. At week 32, patients in the reference aflibercept group are randomized to continue receiving reference product or switched to the biosimilar. The primary endpoint, change in BCVA from baseline at week eight, was achieved with 6.7 letters for SB15 and 6.6 letters for aflibercept, (95% confidence interval [CI], -1.3 to 1.4). Equivalent changes in BCVA and OCT CST were seen up to week 32. No new safety signals were found, and the AEs were similar in incidence and severity between SB15 and reference aflibercept up to week 32. Immunogenicity and pharmacokinetic profiles were also similar.22 The final results at week 52 will soon be available.
MYL-1701P is an aflibercept biosimilar candidate being evaluated in the Phase III clinical trial INSIGHT in DME patients. In that study, 355 patients were randomized to receive either MYL-1701P or the reference product aflibercept. At week eight, the primary endpoint, mean change in BCVA, showed therapeutic equivalence between the drugs. Secondary endpoints included analysis of proportion of patients who gained ≥15 ETDRS letters from baseline and safety outcomes. Similar proportions of patients gained > 5 letters at eight weeks (61.4 percent biosimilar versus 62.8 percent reference aflibercept) and similar rates of ≥10 and ≥15 letters improvement were seen. Safety was demonstrated and no significant differences in antidrug antibodies were seen. Follow up was up to 52 weeks.23
Bevacizumab Biosimilars
There are three bevacizumab approved biosimilars in the United States, all for intravenous oncologic applications.24 It’s important to note that they’re not approved for intraocular use, and it’s not recommended to use them intraocularly. The drugs are bevacizumab-awwb, ABP215 (Mvasi), approved in September 2017; bevacizumab-bvzr (PF-06439535, Zirabev), approved in Jun 2019; and bevacizumab-maly (Almysys), approved in April 2022.
In contrast, bevacizumab-vikg (ONS-5010/Lytenava, Outlook Therapeutics) was developed for intraocular use and is being evaluated for that indication. Three registration trials have been performed for ONS-5010 and several more are in the works. In NORSE ONE, 61 nAMD patients received ONS-5010 or ranibizumab 0.5 mg. Positive efficacy and safety in the first trial led to NORSE TWO, the pivotal Phase III superiority trial for nAMD. In NORSE TWO, 228 patients were randomized to bevacizumab-bvzr mg dosed monthly or ranibizumab 0.5 mg dosed monthly for three months, and then quarterly (PIER dosing regimen) up to month 12. The primary endpoint, the difference in the proportion of patients who gained at least 15 letters in BCVA at 11 months, was reached in 41.7 percent (p=0.0052). NORSE THREE is an open label study that enrolled 197 patients and showed a positive safety profile. NORSE FOUR is planned for BRVO and NORSE FIVE and SIX are planned for DME.25
ONS-5010 has been filed as a new biologic with the FDA instead of as a biosimilar because there’s no FDA approved bevacizumab for ophthalmic indications.
In conclusion, biosimilars offer affordability and accessibility to anti-VEGF therapy as compared to reference anti-VEGFs. This premise requires that ophthalmologists embrace the use of biosimilars. Only time will tell if ophthalmologists indeed choose biosimilars as first-line agents, because of their lower cost and results that are expected to be similar to reference anti-VEGF products. Although the data indeed show equivalence in terms of efficacy and safety, data are only available for up to one year for the approved biosimilars in the United States. More experience with biosimilars will show whether these agents can continue to show safety profiles similar to those of reference anti-VEGFs over the long term. Eventually, with continued demonstration of safety in real-world studies, it’s likely that physicians will become more comfortable with these biosimilars, and their use will increase. In the future, however, the introduction of newer agents that have greater efficacy and durability will compete with biosimilars and existing anti-VEGFs. Until then, biosimilars offer us another option for treating our patients.
Dr. Lim is the Marion H. Schenk Esq., Chair and UIC Distinguished Professor of Ophthalmology and director of the Retina Service at the University of Illinois at Chicago.
Department Editors
Dr. Regillo is the director of the Retina Service of Wills Eye Hospital, a professor of ophthalmology at Thomas Jefferson University School of Medicine and the principle investigator for numerous major international clinical trials.
Dr. Yonekawa is an assistant professor of ophthalmology at Sidney Kimmel Medical College at Thomas Jefferson University. He serves on the Education Committee of the American Society of Retina Specialists and on the Executive Committee for the Vit Buckle Society, where he is also the vice president for academic programming.
1. www.fda.gov. Biological product definitions. P 1-2.
2. https://www.fda.gov/drugs/therapeutic-biologics-applications-bla/biosimilars. Accessed December 6, 2022.
3. Derbyshire M, Shina S. Patent expiry dates for biologicals: 2018 update. GaBI 2019;8:24–31.
4. https://www.gabionline.net/guidelines/US-guidelines-for-biosimilars. Accessed December 6, 2022.
5. Wessels U, Zadak M, Reiser A, Brockhaus J, Ritter M, Abdolzade-Bavil A, Heinrich J, Stubenrauch K. Immunogenicity testing of therapeutic antibodies in ocular fluids after intravitreal injection. Bioanalysis 2018;10:11:803-814.
6. https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval. Accessed December 6, 2022
7. Sharma A, Kumar N, Bandello F, et al. Need of education on biosimilars amongst ophthalmologists: Combating the nocebo effect. Eye 2020;34:1006–1007.
8. Kristensen LE, Alten R, Puig L, Philipp S, Kvien TK, et al. Non-pharmacological effects in switching medication: The nocebo effect in switching from originator to biosimilar agent. BioDrugs 2018;32:397-404.
9. Kapur M, Nirula S, Naik MP. Future of anti-VEGF: Biosimilars and biobetters. Int J Retin Vitr 2022;8:2. https://doi.org/10.1186/s40942-021-00343-3.
10. Sharma A, Kumar N, Parachuri N, et al. Biosimilars for retinal diseases: An update. Am J Ophthalmol 2021;224:36–42.
11. Sameera VV, Ayachit A, Joshi S, Guruprasad AS. Safety and efficacy of Razumab—the new biosimilar in India: Our experience. Kerala J Ophthalmol 2016;28:180.
12. Sharma S, RE-ENACT Study Investigators Group, Khan MA, Chaturvedi A. Real-life clinical effectiveness of Razumab (world’s first biosimilar ranibizumab) in wet age-related macular degeneration, diabetic macular edema, and retinal vein occlusion: A retrospective pooled analysis. Int J Ophthalmol Eye Res 2018;6:4:377–83.
13. Sharma S, Khan MA, Chaturvedi A, RE-ENACT Study Investigators Group. Real life clinical effectiveness of Razumab (World’s First Biosimilar Ranibizumab) in wet age-related macular degeneration: A subgroup analysis of pooled retrospective RE-ENACT study. Int J Ophthalmol Eye 2018;6:2:368–373.
14. Sharma S, Khan MA, Chaturvedi A, RE-ENACT Study Investigators Group. Real life clinical effectiveness of Razumab (world’s first biosimilar ranibizumab) in retinal vein occlusion: A subgroup analysis of pooled retrospective RE-ENACT study. Ophthalmologica 2018;1–8:26.
15. Verma L, Thulasidas M, Purohit A, Gupta A, Narula R, Talwar D. Clinical efficacy and safety of Razumab (CESAR) study: Our experience with the world’s first biosimilar Ranibizumab. Indian J Ophthalmol 2021;69:2:347–51.
16. Sharma A, Kumar N, Kuppermann B, Francesco B, Lowenstein A. Ophthalmic biosimilars: Lessons from India. Indian J Ophthalmol 2019;67:1384.
17. Sharma S, Sharma T, Prasad S, Gopalakrishnan M, Chaturvedi A. Treatment landscape of macular disorders in Indian patients with the advent of Razumab (world’s first biosimilar ranibizumab): A comprehensive review. Ophthalmol Ther 2021;10:3:431-443.
18. Woo SJ, Veith M, Hamouz J, et al. Efficacy and safety of a proposed ranibizumab biosimilar product vs a reference ranibizumab product for patients with neovascular age-related macular degeneration: A randomized clinical trial. JAMA Ophthalmol 2021;139:1:68-76.
19. Bressler NM, Veith M, Hamouz J, Ernest J, Zalewski D, Studnička J, Vajas A, Papp A, Vogt G, Luu J, Matuskova V, Yoon YH, Pregun T, Kim T, Shin D, Oh I, Jeong H, Kim MY, Woo SJ. Biosimilar SB11 versus reference ranibizumab in neovascular age-related macular degeneration: 1-year phase III randomised clinical trial outcomes. Br J Ophthalmol 2021 Oct 16:bjophthalmol-2021-319637. doi: 10.1136/bjophthalmol-2021-319637. Epub ahead of print. PMID: 34656987.
20. CIMERLI (ranibizumab-eqrn) U.S. Prescribing Information, August 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761165s000lbl.pdf. Accessed December 6, 2022.
21. Holz FG, Oleksy P, Ricci F, et al. Efficacy and safety of biosimilar FYB201 compared with ranibizumab in neovascular age-related macular degeneration. Ophthalmology 2022;129:1:54-63.
22. Woo SJ, Sadda SR, Bradvica M, et al. SB15, a proposed biosimilar to aflibercept, in nAMD: 32-week results. Presented at: AAO 2022; September 30-October 3, 2022; Chicago. Poster PO381.
23. Bressler SB, Barve A, Beckmann K, et al. MYL-1701P (proposed biosimilar aflibercept) compared to Eylea in DME: Outcomes from the phase 3 INSIGHT study. Presented at: AAO 2022; September 30-October 3, 2022; Chicago. Poster PO387.
24. https://www.onclive.com/view/fda-approves-third-bevacizumab-biosimilar. Accessed December 6, 2022.
25. https://outlooktherapeutics.com/lytenava-clinical-progress/. Accessed December 6, 2022.