Development
Ocular development begins with the formation of the optic vesicle from the neuroectoderm largely under the influence of the Sonic hedgehog (SSH), PAX2 and PAX6 genes. The optic vesicle and subsequent growth of the eye influences the formation of the surrounding orbital soft tissue contents and the bony orbital walls.
Various developmental genes regulate tissue interactions during embryogenesis, including fibroblast growth factor receptors (FGFR) 1, 2 and 3, and Twist homologue 1 (TWIST1), and the transforming growth factor, beta receptor 1 (TGFBR1). Awareness of orbital, ocular and adnexal development and the migratory pattern of neural crest cells is useful for understanding the etiology of congenital orbital, eyelid and lacrimal anomalies. The typical location of dermoid cysts at the frontozygomatic and frontoethmoidal suture lines is the result of a sequestration of surface ectoderm in areas of neural crest cell fusion. The superficial spread and deep invasion of basal cell carcinoma on the midface has been attributed to the location of the embryonic fusion planes.
Genetic Anomalies of the Eyelids
The genetic forms of ptosis are summarized in Table 1.
• Congenital ptosis. Congenital ptosis resulting from a localized dysgenesis of the levator muscle can occur in isolation or in combination with other malformations. Isolated congenital ptosis is usually not heritable. A few reports indicate the possibility of dominant inheritance and linkage to 1p34.1-p32, or Xq24-q27.1.1,2 The ZFH4 gene (8q21.1) may be a candidate gene.3
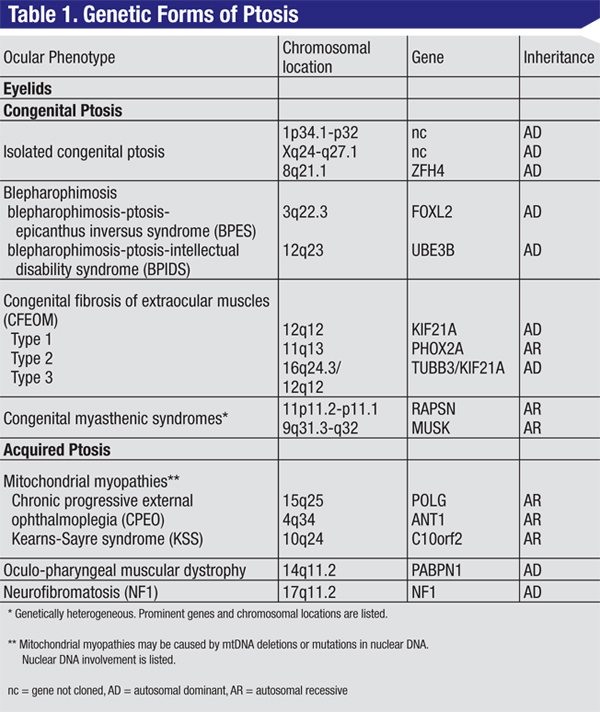 |
Autosomal-dominant blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) results from mutation or deletion of the FOXL2 gene (3q22.3; See Figure 1). BPES1 is associated with premature ovarian failure and infertility in girls. BPES2 is not associated with ovarian failure, and can be transmitted by both males and females. Intellectual disability in patients with BPES may be due to larger deletions involving 3q22.3, or part of the blepharophimosis-ptosis-intellectual disability syndrome (BPIDS) caused by heterozygous mutations in the UBE3B gene (12q23).6
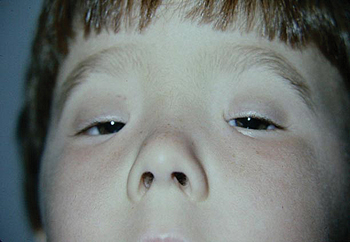
Congenital fibrosis of the extraocular muscles (CFEOM) types 1, 2 and 3 are characterized by ptosis and complete ophthalmoplegia or other restricted eye movements. CFEOM is considered a congenital cranial dysinnervation disorder (CCDD) and is believed to result from aberrant innervation of the extraocular muscles. In CFEOM1, the primary position of both eyes is below the horizontal midline with severe restriction of elevation of either eye above the midline. Horizontal movements of the eyes range from normal to severely restricted. Marcus Gunn jaw wink may be present. The condition is autosomal dominant and results from heterozygous mutations in KIF21A (12q12). CFEOM2 is an autosomal recessive disorder characterized by congenital bilateral exotropic ophthalmoplegia and ptosis, with pupillary abnormalities, in particular miosis. It is due to mutations in PHOX2A (11q13). CFEOM3 patients manifest with ptosis and ophthalmoplegia, and typically demonstrate a broader variation in phenotype than CFEOM1 and CFEOM2 patients. The CFEOM3 phenotype is genetically heterogeneous and may be caused by mutations in the TUBB3 (16q24.3) or KIF21A genes. Patients with CFEOM3 may also have facial dysmorphism, cognitive impairment, thin corpus callosum or digital anomalies.
Congenital myasthenic syndromes are genetic disorders of the neuromuscular junction. Patients manifest with ptosis and ophthalmoplegia, easy fatiguability, and facial, bulbar, neck and limb weakness, or respiratory insufficiency. They are associated with mutations in different genes encoding proteins involved in presynaptic, synaptic or postsynaptic neuromuscular transmission, including the RAPSN gene (11p11.2-p11.1), which plays an essential role in the clustering of acetylcholine receptors at the endplate, and the muscle-specific protein kinase (MUSK, 9q31.3-q32) gene, critical for synaptic differentiation.
• Acquired ptosis. Ptosis with reduced levator function and/or ophthalmoplegia often points to a myogenic cause. The differential diagnosis includes mitochondrial myopathies, oculopharyngeal muscular dystrophy (OPMD), oculopharyngodistal myopathy (ODM) and myotonic dystrophy.
|
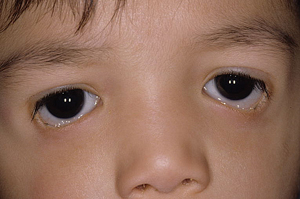
OPMD is characterized by progressive ptosis, external ophthalmoplegia, dysphagia and proximal limb weakness with onset usually in the sixth decade, and is caused by heterozygous mutations in PABPN1 (14q11.2). ODM is characterized by ptosis, masseter, facial, bulbar muscle and distal limb weakness beginning usually in the patient’s 40s. The genetic defect causing ODM has not been elucidated. Myotonic dystrophy is the most common adult-onset muscular dystrophy. Ocular findings include ptosis, ophthalmoplegia, hypotony and polychromatic cataracts. Patients may have frontal alopecia, cardiomyopathy and testicular atrophy. It is caused by expansion of a heterozygous trinucleotide repeat (CTG)n in the DMPK gene (19q13).
Ptosis may also be due to eyelid involvement by nodular or plexiform neurofibromas in neurofibromatosis, an autosomal dominant condition characterized by extreme variability of expression both within and among families. The neurofibromin protein, encoded by the NF1 gene (17q11.2) is a tumor suppressor gene. Plexiform neurofibroma characteristically produces an S-shaped contour to the eyelid which when palpated may feel like “a bag of worms.” Orbital involvement in type 1 neurofibromatosis includes optic nerve glioma, other orbital tumors such as neurilemmoma and meningioma, or a defect in the greater wing of the sphenoid bone which can result in pulsating proptosis as the intracranial contents make contact with orbital tissues.
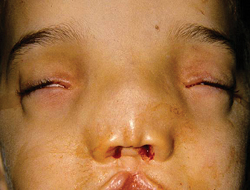
• Eyelid coloboma. Eyelid coloboma may be isolated or associated with facial abnormalities.7 Upper-lid coloboma may be seen in the oculoauriculovertebral spectrum (OAVS), including the subtype known as Goldenhar syndrome. OAVS, due to abnormal development of the first and second branchial arch, is associated with multiple loci and one known gene, SALL1 (16q12.1) and may be inherited in an autosomal dominant manner.8
|
• Other eyelid anomalies. Lymphedema-distichiasis is an autosomal dominant disorder caused by mutations in the FOXC2 gene (16q24.1), and presents as distichiasis of upper and lower eyelids and ptosis, with lymphedema of the limbs.
Euryblepharon, characterized by horizontal enlargement of the palpebral fissure with associated lateral canthal malposition and lateral ectropion, may be seen in association with genetic syndromes such as the Niikawa-Kuroki (formerly Kabuki Makeup) syndrome and blepharo-cheilo-dontic syndrome (BCDS). Niikawa-Kuroki syndrome is a congenital mental retardation syndrome with postnatal short stature, facial dysmorphism, a cleft or high-arched palate and skeletal abnormalities of the vertebrae, hands and hip joints. It is caused by heterozygous mutations in the MLL2 gene (12q12-q14).9 BCDS is also a rare autosomal-dominant disorder characterized by upper eyelid distichiasis, euryblepharon, bilateral cleft lip and palate and conical teeth. No specific gene or locus has been identified.
Congenital ectropion is rare and often caused by a vertical deficiency of the anterior lamella of the eyelids. It may be associated with genetic disorders such as blepharophimosis syndrome, Down syndrome or ichthyosis (collodion baby). Congenital ichthyosis is a heterogeneous group of disorders of keratinization characterized primarily by abnormal skin scaling over the whole body. Collodion babies have a taut, shiny, translucent or opaque membrane that encases the entire body and lasts for days to weeks. Congenital ichthyosis is autosomal recessive and genetically heterogeneous. Mutations in the TGM1(14q12) gene account for majority of cases.
Basal cell nevus syndrome also known as Gorlin-Goltz syndrome or nevoid basal cell carcinoma (BCC) syndrome, is caused by mutations in the PTCH1 (9q22), PTCH2 (1p32) or SUFU genes (10q24-q25). It is an autosomal-dominant cancer predisposition syndrome resulting in multiple BCCs. Developmental malformations, hamartomas and dysplastic lesions are consistent and striking components of the nevoid BCC syndrome. Clinically, diagnosis of nevoid BCC syndrome is made in the presence of two major or one major and two minor criteria (See Table 2).10
Genetic Anomalies: Orbit & Globe
• Craniosynostosis. The craniosynostoses are a group of disorders characterized by premature fusion of one or more of the cranial sutures resulting in restriction of growth in a direction parallel to the orientation of the fused suture. The bony orbit is often shallow with recession of the orbital rims resulting in exorbitism.
|
• Facial clefts. Facial clefts are congenital anomalies that occur due to failure of fusion of normal embryonic clefts or facial processes. Clefts are defined according to the system of Tessier based on their position, and are grouped into midline, paramedian, orbital, or lateral clefts. They may be associated with cranial anomalies, mostly encephalocele. Frontonasal dysplasia (FND) due to median facial clefting is characterized by ocular hypertelorism, a broad nasal root, unilateral or bilateral clefting of the alae nasi, anterior cranium bifidum occultum, and a V-shaped or widow’s peak frontal hairline (See Figure 3). FND is genetically heterogeneous and may be caused by homozygous mutations in the ALX3 gene (1p13.3), ALX4 gene (11p11.2) or ALX1 gene (12q21.3-q22). A type of FND, frontorhiny, is characterized by distinctive features of FND with upper eyelid ptosis and midline dermoid cysts of craniofacial structures.11
• Anophthalmia/Microphthalmia. True anophthalmia refers to complete absence of the globe. It occurs when the neuroectoderm of the primary optic vesicle fails to develop properly from the anterior neural tube. Microphthalmia is defined as a globe with a total axial length that is at least two standard deviations below the mean for age. Both occur secondary to developmental arrest at various stages of growth of the optic vesicle. Severe microphthalmia may present as clinical anophthalmia with globe remnants only visible by ultrasound or by histology. As the development of the orbital region, as well as the lids and the fornices, is dependent on the presence of a normal-sized eye in utero, anophthalmia and microphthalmia result in secondary underdevelopment of the orbit, lids, and socket. Anophthalmia and microphthalmia may occur unilaterally or bilaterally, in isolation or in association with systemic disease. Genetic causes include chromosomal aberrations (e.g. trisomy 13 and 18), mutations or deletions involving the SOX2, SIX6, STRA6, HESX1, BCOR, SHH, PAX6 and RAX genes, and syndromes such as Goltz, Meckel-Gruber, Seckel, cerebro-oculo-nasal, branchio-oculo-facial, Walker-Warburg, and CHARGE syndromes, among others.12
• Cryptophthalmos. Complete cryptophthalmos is characterized by absence of the eyelids, which are replaced by skin extending in continuity with the cheek over the orbit. A new grading of upper eyelid colobomas and cryptophthalmos suggests that the two entities may represent opposite ends of the same eyelid malformation spectrum.7 Cryptophthalmos may be isolated or syndromic.
|
Lacrimal Gland, Drainage System
• Congenital alacrimia. Autosomal dominant aplasia of the lacrimal glands with or without aplasia of the salivary glands is a rare condition characterized by dry eye and, in the latter case, xerostomia. It is caused by mutations in the gene encoding FGF10 (5p12) although two other loci are known.14 Lacrimoauriculodentodigital syndrome (LADD), also known as Levy-Hollister syndrome, is an allelic disorder with a more severe phenotype.15 Besides alacrimia and xerostomia, it is characterized by malformation of the external ears, teeth (unerupted and dysplastic teeth), kidneys (renal agenesis, nephrosclerosis) and ditory system (malformation of the auricles (auricular dysplasia, congenital hearing loss). Allgrove syndrome, also known as Triple-A syndrome or Achalasia-Addisonianism-Alacrimia syndrome, is a rare, autosomal recessive disorder. It is caused by mutations in the AAAS gene (12q13.13). Patients with this condition may also have autonomic dysfunction such as pupillary abnormalities, abnormal reaction to intradermal histamine, abnormal sweating and orthostatic hypotension. Another condition associated with defective lacrimation and autonomic dysfunction is familial dysautonomia (Riley-Day syndrome), an autosomal recessive disorder caused by mutations in the IKBKAP gene (9q31).
• Nasolacrimal drainage system anomalies. Congenital nasolacrimal duct obstruction is a common disorder that may be associated with many genetic disorders, such as branchiooculofacial syndrome (TFAP2A gene mutations, 6p24.3), trisomy 21 (Down syndrome), Johanson-Blizzard syndrome (UBR1 gene mutations, 15q13-21.1), dyskeratosis congenita (DKC1 gene mutations, Xq28) and the Treacher-Collins-Franceschetti syndrome. Lacrimal sac fistula is seen in Down syndrome. Patients with Down syndrome may also experience epiphora due to floppy lid syndrome, congenital ectropion or hypotonia of the pump mechanism. REVIEW
Dr. Ganesh is a consultant in Pediatric Ophthalmology and Ocular Genetics, and Dr. Al-Mujaini a senior consultant in Oculoplastics, both at the Sultan Qaboos University Hospital, Muscat, Oman. Dr. Levin is chief of Pediatric Ophthalmology and Ocular Genetics at Wills Eye Institute, 840 Walnut St., Ste. 1210, Philadelphia, PA 19107-5109. Phone: (215) 928-3914; fax: (215) 928-3983; e-mail: alevin@willseye.org.
1. Engle EC, Castro AE, Macy ME, et al. A gene for isolated congenital ptosis maps to a 3 cM region within 1p32-p34.1. Am J Hum Genet 1997;60:1150-7.
2. McMullanTFW, Collins AR, Tyers AG, et al. A novel X-linked dominant condition: X-linked congenital, isolated ptosis. Am J Hum Genet 2000;66:1455-60.
3. McMullan TFW, Crolla JA, Gregory SG, et al. A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced translocation. Hum Genet 2002;110:244-50.
4. Ganesh A, Al-Kindi A, Jain R, Raeburn S. The phenotypic spectrum of Baraitser - Winter Syndrome: A new case and review of literature. JAAPOS 2005;9:604-6.
5. Pao KY, Levin AV. Genetics in Oculoplastics. In: Black EH, Nesi FA, Gladstone GJ, Levine MR, eds. Smith and Nesi’s Ophthalmic Plastic and Reconstructive Surgery. 3rd ed. New York: Springer; 2012:1249-64.
6. Basel-Vanagaite L, Dallapiccola B, Ramirez-Solis R, et al. Deficiency for the ubiquitin ligase UBE3B in a blepharophimosis-ptosis-intellectual-disability syndrome. Am J Hum Genet 2012;91:998-1010.
7. Nouby G. Congenital upper eyelid coloboma and cryptophthalmos. Ophthal Plast Reconstr Surg 2002;18:373-7.
8. Vendramini-Pittoli S, Kokitsu-Nakata NM. Oculoauriculovertebral spectrum: Report of nine familial cases with evidence of autosomal dominant inheritance and review of the literature. Clinical Dysmorphology 2009;18:67-77.
9. Li Y, Bogershausen N, Alanay Y, et al. A mutation screen in patients with Kabuki syndrome. Hum Genet 2011;130:715-24.
10. Honavar SG, Shields JA, Shields CL, Eagle RC Jr, Demirci H, Mahmood EZ. Basal cell carcinoma of the eyelid associated with Gorlin-Goltz syndrome. Ophthalmology 2001;108:1115-23.
11. Twigg SRF, Versnel SL, Nurnberg G, et al. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet 2009;84:698-705.
12. Bardakjian T, Weiss A, Schneider AS, editors. Anophthalmia/Microphthalmia Overview. [monograph on the internet] Gene Reviews; 2006 [cited 2013 Aug 16]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1378/
13. Vogel MJ, van Zon P, Brueton L, et al. Mutations in GRIP1 cause Fraser syndrome. J Med Genet 2012;49:303-6.
14. Entesarian M, Matsson H, Klar J, et al. Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nature Genet 2005;37:125-8.
15. Milunsky JM, Zhao G, Maher TA, Colby R, Everman DB. LADD syndrome is caused by FGF10 mutations. Clin Genet 2006;69:349-54.


