Paraneoplastic syndromes occur in as many as 10 to 15 percent of cancer patients.1 Those involving the visual system are considerably less common, and can be easily overlooked given their subtle warning signs. Here we review the retinal-based paraneoplastic syndromes, specifically cancer-associated retinopathy, melanoma-associated retinopathy (a lesser-known entity), paraneoplastic vitelliform maculopathy and bilateral diffuse uveal melanocytic proliferation.
Cancer-Associated
Approximately 100 cases of cancer-associated retinopathy have been reported in the literature, yet CAR is the most prevalent paraneoplastic retinopathy. Nearly half of these patients presented with visual symptoms prior to the diagnosis of their underlying malignancy.1 The primary tumor accounting for the vast majority of CAR cases is small-cell lung carcinoma, followed by gynecologic (ovarian, endometrial and cervical) and breast malignancies.2 Less commonly, non-small-cell lung cancer, lymphoma, prostate and bladder cancers have been reported.2
In a recent review of 55 patients, average age of onset was 65 years, with men and women being equally affected.2 Symptoms usually present bilaterally, over the course of weeks to months. Patients typically complain of photopsias, severely reduced visual acuity, color impairment, photosensitivity, central or peripheral ring scotomas and nyctalopia, reflecting involvement of both rod and cone photoreceptors. A lesser-known variant of CAR, termed cancer-associated cone dysfunction, characteristically affects only cones.3,4,5
Fundus examination in CAR is often unremarkable; however, advanced findings may include any combination of optic nerve pallor, attenuated retinal vasculature, retinal pigment epithelial changes and vitreous cells.2,6 Retinal pigment dispersion is typically absent or sparse in distribution.31 The electroretinography findings in CAR are illustrative of global retinal dysfunction, with severely reduced, if not extinguished, scotopic and photopic a- and b-waves.7
On an immunologic basis, molecular mimicry has been proposed as the pathogenic mechanism behind the disease. By this process, tumor cells expressing antigenic epitopes with retinal photoreceptors elicit an immune response that cross-reacts with the retina, disrupting normal signal transmission. The two most commonly identified retinal antigens associated with CAR are recoverin and α-enolase.2,7,8,9 Numerous other antigens continue to be implicated, including transducin, arrestin, tubby-like protein 1 and photoreceptor cell-specific nuclear receptor.
While no effective treatment for CAR exists, long-term immunosuppression remains the mainstay of therapy. Some benefit has been suggested from combinations of corticosteroids, plasmapheresis and intravenous immunoglobulin (IVIG), which presumably decrease circulating anti-retinal antibodies and help to stabilize VA. Treatment of the underlying malignancy does not improve vision.2,7,8 Overall, the visual prognosis is poor, and rapid, relentless vision loss may occur.
Melanoma-Associated
Unlike CAR, which frequently precedes the discovery of systemic malignancy, patients with melanoma-associated retinopathy (MAR) usually carry an existing diagnosis of melanoma. With a latency period averaging 3.6 years from diagnosis of the primary neoplasm (ranging two months to 19 years), the onset of MAR often heralds the presence of nonocular metastasis.10 All but two of the 64 previously reported cases of MAR were associated with cutaneous melanomas; however, a recently published series of 11 patients also implicates choroidal and ciliary body melanomas and even choroidal nevi.11 Age of onset is commonly during the sixth decade of life, with a significant male predilection with a male to female ratio of 4.7:1.10
Clinical features typically reflect rod-mediated dysfunction, with patients experiencing sudden onset of shimmering photopsias and nyctalopia, without the degree of VA or color impairment seen with CAR. However, similar to CAR, initial fundus examination is often normal. The aforementioned posterior pole changes seen with disease progression in CAR have also been described in MAR.10 Visual field testing most commonly reveals generalized constriction. Arcuate defects, and central and paracentral scotomas have also been reported.10 The ERG in MAR is classically electronegative, demonstrating a preserved dark-adapted a-wave, indicating normal photoreceptor function, followed by a markedly attenuated b-wave, reflecting either bipolar cell dysfunction or disruption of transmission to the bipolar cell.10
It has been postulated that autoantibodies directed against the postsynaptic receptors of the depolarizing, or “ON” bipolar cells, disrupt neural transmission with associated rod photoreceptors, resulting in the predominantly rod-related visual compromise.2,12,13 Attempts to elucidate a specific antigen of these anti-bipolar cell antibodies have yielded numerous candidates, the clinical utility of which is addressed later.11
Treatment for MAR has been generally ineffective, with successes limited to individual cases. Cytoreductive therapy with surgery or radiation to the primary malignancy followed by adjuvant immunotherapy with IVIG to decrease tumor burden has been proposed as one treatment strategy.10 Other combinations of corticosteroids, plasma exchange and IVIG leading to improvement have been reported in the literature. Overall visual prognosis is guarded, and while MAR is traditionally believed to remain stable and non-progressive, a minority of cases do lose central vision.10
|
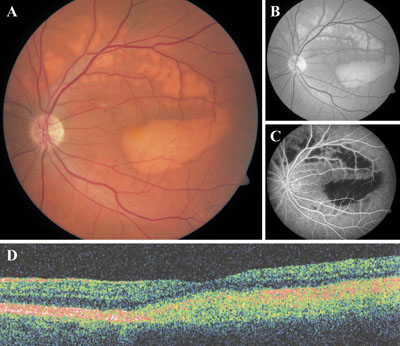
| Figure 1. Paraneoplastic vitelliform maculopathy in a 58-year-old man with a history of metastatic cutaneous melanoma to the axillary lymph nodes who presented with mild blurring of vision in the left eye (20/50) and preserved acuity (20/20) in the right eye. Color fundus photography (A, only left eye pictured) was remarkable for bilateral, multiple, subretinal, vitelliform lesions of the macula. B) Red-free images of the fundus lesions are also presented. C) Fluorescein angiography demonstrated predominant blockage of background fluorescence by the material, which appeared to layer and gravitate into the inferior macula. D) Optical coherence tomography confirmed these infiltrates to be at the level of the RPE without associated serious fluid. Visual field and ERG were essentially normal, autoantibodies to bipolar cells were not detected, and EOG revealed only a mildly decreased Arden ratio to 1.7. The patient developed metastatic spread to his small bowel two months later.
(Image courtesy Peter J. Keters, MD) |
In recent years, a growing number of reports have emerged of melanoma patients developing a presumed paraneoplastic retinopathy with features uncharacteristic of typical MAR.14-22 To our knowledge, at least 14 cases exist in the current literature, which we will refer to as paraneoplastic vitelliform maculopathy (PVM). Both choroidal and cutaneous melanomas have been associated, and the incidence of metastases in conjunction with onset of the ocular phenomena has varied. A unifying feature of these cases is the presence of vitelliform lesions in the posterior pole at the level of the RPE with associated serous macular detachment, features not previously attributed to MAR (See Figure 1).14-21 Symptoms at onset are diverse, including nyctalopia and photopsias, as previously described in MAR, as well as central vision loss and field deficits secondary to the macular pathology. Results of anti-retinal antibody and ERG testing have been variable.
Clinically, PVM may more closely resemble other vitelliform dystrophies, such as acute exudative polymorphous vitelliform maculopathy (AEPVM), or Best macular dystrophy (BMD). Indeed, in a recent case report, a patient presented with the classic findings of AEPVM, as first described by Donald Gass, MD, of bilateral macular and peripapillary subretinal yellowish lesions arranged in a honeycomb-like pattern that hyperfluoresce early and stain late with fluorescein angiography.20,23-25 There were associated serous macular detachments with gradual gravitation of the vitelliform material to the inferior macula later in the disease course. This patient was diagnosed four months later with metastatic melanoma of unknown primary origin. Successful systemic treatment of the metastatic disease correlated with improvement of numerous visual parameters, including VA as well as anatomic resolution of macular subretinal fluid bilaterally. The authors additionally demonstrated antibodies in the patient’s serum to peroxiredoxin 3 (PRDX3), an RPE peroxidase, suggesting the underlying pathogenesis may be an autoimmune-mediated retinal pigment epitheliopathy.20
In a separate case, a patient with a remote history of choroidal melanoma status post-enucleation developed a serous macular detachment seen as a vitelliform “pseudohypopyon” lesion in the contralateral eye within one year of metastatic spread to the liver and lungs. ERG was within normal limits, while electro-oculography showed a pathological Arden ratio of 1.1, consistent with Best macular dystrophy. However, no family history of BMD was elicited, and genetic analysis for VMD2 mutation was negative. Furthermore, the investigators detected the presence of circulating autoantibodies against bestrophin, which they postulated to be paraneoplastic in nature.21
Whether or not these cases represent an atypical variant of MAR or a unique entity is a subject that welcomes further attention.
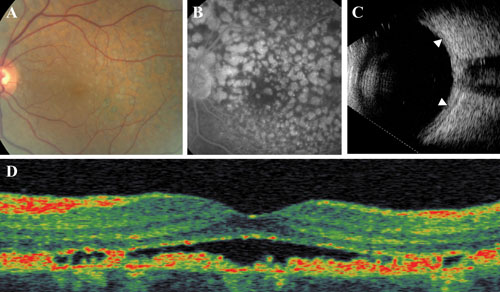 |
| Figure 2. Diffuse uveal melanocytic proliferation (DUMP) in a 55-year-old woman with a two-year history of metastatic, small-cell lung carcinoma who presented with decreased visual acuity in the left eye (20/40). She was noted to have a moderate nuclear sclerotic cataract in that eye. A) Color fundus photography revealed islands of atrophic retinal pigment epithelium separated by a reticular pattern of yellow-orange pigmentation in the left macula. Corresponding fluorescein angiography (B) was remarkable for nummular areas of early hyperflourescence consistent with RPE atrophy. C) B-scan ultrasound revealed extensive choroidal thickening of the affected left eye (arrowheads). D) OCT of the same eye demonstrated neurosensory detachment over the macula with focal areas of RPE atrophy and hypertrophy. The respective studies in the right eye were all normal.
(Image courtesy Shartan Reddy, MD) |
Since first being described by Robert Machemer, MD, more than 40 years ago in a patient with pancreatic cancer,26 there have been 40 published cases of bilateral diffuse uveal melanocytic proliferation (BDUMP). This rare disorder usually presents between the sixth and ninth decades of life with rapid onset and painless, bilateral vision loss accompanied by subtle ophthalmoscopic findings. Dr. Gass and colleagues described the five cardinal signs of BDUMP: 1) multiple, subtle, round, red patches in the fundus; 2) multifocal early hyperfluorescence of these patches reflecting window defects from overlying RPE atrophy; 3) diffuse thickening of the choroid with focally elevated pigmented tumors; 4) exudative retinal detachments; and 5) rapidly progressive cataract formation.27 An orange, polygonal “giraffe pattern” of the fundus, secondary to RPE accumulation bordering zones of pigment epithelial atrophy, is also characteristic.38 Though bilateral by definition, a unilateral case of DUMP has been reported (See Figure 2).39
BDUMP often precedes the diagnosis of the systemic malignancy, typically involving the genital tract in women (ovarian or uterine carcinomas) and lung or pancreatic cancer in men.28,29 Through unknown mechanisms, the primary tumor stimulates the proliferation of uveal melanocytes. A popular theory centers around the production of melanocytic growth factors by tumor cells, with subsequent release into the circulation. In agreement with this hypothesis is the prevalence of skin or mucous membrane pigmented lesions in BDUMP patients (up to 26 percent in one review).28,30 In addition to the melanocytic proliferation, Dr. Gass hypothesized that other toxic or immunologic factors must also participate in the disease pathogenesis, contributing to the degeneration of the RPE, retina and lens.27 Debate exists as to whether these changes occur as a by-product of increased metabolic demands and ensuing hypoxia from the replicating melanocytes or as a distinct paraneoplastic process independent of the melanocytic proliferation.
Ultimately, visual prognosis in BDUMP is poor, with progressive deterioration of vision to near complete blindness. Surgical intervention for cataracts and exudative retinal detachment are unsuccessful. Patients often die within a year of diagnosis secondary to the systemic tumor burden.
Diagnostic Dilemmas
No definitive diagnostic criteria exist for the paraneoplastic retinopathies. Diagnosis, rather, is made by combining clinical features, examination findings and ancillary testing results into a cohesive rationale. Of critical importance, however, is to maintain a high index of suspicion, which may be challenging, given their rare occurrence and the possibility that their onset may precede any knowledge of a systemic malignancy. In a patient with suspected CAR and no known history of malignancy, appropriate workup includes a detailed history and physical examination, and chest X-ray.31 Additional imaging studies may be warranted depending on findings of the initial workup.
One must also entertain alternative retinal degenerative diagnoses: acute zonal occult outer retinopathy; retinitis pigmentosa; nonparaneoplastic autoimmune retinopathy; cone dystrophy; toxic retinopathy (chloroquine, hydroxychloroquine, thioridazine); syphilis and avitaminosis A.
Warranting further attention is acute zonal occult outer retinopathy (AZOOR), an idiopathic retinal disease characterized by variable degeneration of photoreceptors. Like the paraneoplastic retinopathies, AZOOR is rare, with 131 cases reported to date.32 Overlapping features among these entities can be cause for confusion. AZOOR typically affects young (average age 36.7 years) Caucasian (89 percent of patients) women (male:female ratio of 1:3.2).32 In contrast, paraneoplastic retinopathies most commonly present during the sixth to seventh decades of life, show no racial preference, and with the exception of MAR, which affects men more than women, have no gender predilection.
Dr. Gass noted a defining feature of AZOOR to be the acute onset of decreased vision in a zone of visual field, accompanied by photopsia.33 In fact, VA is minimally affected in AZOOR (Vision of 20/40 or better in 74 percent of eyes),32 similar to MAR and in stark contrast to CAR. Furthermore, while paraneoplastic retinopathies are classically bilateral at onset, AZOOR has been noted to present either unilaterally (62 percent of patients), or bilaterally (39 percent). Fundus examination on presentation is usually unremarkable in AZOOR, MAR and CAR. Over time in AZOOR, RPE mottling analogous to retinitis pigmentosa can develop in zones of visual field loss. Retinal vascular abnormalities, however, are rare in AZOOR,32 unlike the vascular attenuation that has been described in both RP and paraneoplastic retinopathies. In contrast to RP, intraretinal bone spicule migration is rare with either AZOOR or paraneoplastic retinopathies. The presence of cystoid macular edema, epiretinal membrane or posterior subcapsular cataract formation is more suggestive of RP.
Additional testing, notably full-field ERG, can be invaluable in diagnosing and distinguishing amongst AZOOR, paraneoplastic and other retinal degenerations. In AZOOR, an abnormal ERG is demonstrated in the vast majority of cases. It is interesting to note that despite the focal nature of AZOOR clinically, its ERG changes depict a more global retinal dysfunction. The most consistent abnormality noted in a series of 28 patients was a markedly delayed implicit time of the 30-Hz cone flicker response.34 Moderate reductions in a- and b-wave amplitudes are also common, but not to the extent of the severely depressed to extinguished waveforms described in CAR. Additionally, the electronegative ERG pattern of MAR is not typical of AZOOR.32
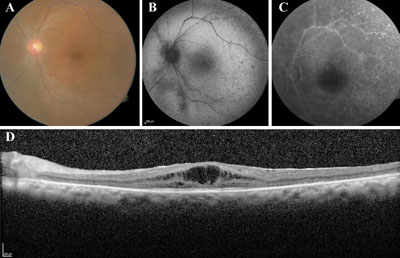 |
| Figure 3. A 53-year-old woman referred for rapidly declining vision in both eyes. A) Color fundus photography (only left eye pictured) revealed marked attenuation of the arteroiles, and diffuse RPE changes bilaterally. B & C) Autoflourescence imaging showed patchy areas of absent autoflourescence suggestive of RPE atrophy, which was supported by the hyperfluorescent window defects seen on fluorescein angiography. D) OCT imaging displayed cystoid macular edema bilaterally, which did not leak on FA. Visual fields were severely constricted, and ERG was extinguished. Anti-retinal antibody testing was positive for antibodies to alpha-enolase; however, a comprehensive systemic workup for malignancy was negative. The patient was diagnosed with RP sine pigmento, and started on oral acetazolamide, which significantly improved the CME. |
It has been previously stated that a diagnosis of CAR or MAR requires the demonstration of anti-retinal antibodies by Western blot, and in the case of CAR, is further confirmed by the presence of anti-recoverin or anti-a-enolase antibodies. Tests for these antibodies are now commercially available (Recombx anti-recoverin test from Athena Diagnostics) and offered at several academic institutions (e.g., University of California at Davis, Ophthalmology Research Laboratories, and Casey Eye Institute, Ocular Immunology Lab). With these recent developments, as well as the myriad of new autoantibodies directed against retinal antigens continuously being reported, clinicians are encouraged to interpret such test results with caution. Demonstration of anti-retinal antibodies, while necessary, is not sufficient alone for diagnosing paraneoplastic retinopathies, since such antibodies have been frequently described in normal human sera. In one study, 57 of 92 (62 percent) of normal serum samples had detectable anti-retinal antibody activity by Western blot.37 Furthermore, positive titers against recoverin have been recovered in cancer patients without any evidence of paraneoplastic retinopathy, and, conversely, in some cases of presumed CAR, the antibodies are not identifiable.35-37
The previous findings raise an important concern, echoed by Lee Jampol, MD, in a recent editorial.40 Given their seemingly ubiquitous nature and increasing number, are many of these retinal antibodies merely epiphenomena, coincidentally present yet not causally related to the disease pathogenesis? If so, our challenge moving forward will be to differentiate the pathogenic and disease-causing antibodies from the benign “innocent bystanders” if we hope to improve our diagnostic yield and add clarity to an elusive group of diseases that are challenging to both diagnose and treat.
Dr. Rahimy is at Jules Stein Eye Institute, UCLA, and the Greater LA VA Healthcare Center. Dr. Sarraf is in the Retinal Disorders and Ophthalmic Genetics Division at Jules Stein Eye Institute, and and the Greater LA VA Healthcare Center.
Notes on images. Figure 1: Reproduced from Paraneoplastic vitelliform retinopathy in metastatic cutaneous melanoma. Krema H, Simpson R, Altomare F, Ebadi M, volume 4, 246-50, 2010 with permission from Wolters Kluwer Health Publishing Group Ltd. Figure 2: Reproduced from Unilateral diffuse uveal melanocytic proliferation (DUMP). Reddy S, Finger PT, volume 91, 1726-7, 2007 with permission from BMJ Publishing Group Ltd.
1. Arnold AC, Lee AG. Systemic disease and neuro-ophthalmology: Annual update 2000. J Neuroophthalmology 2001;21:46-61.
2. Chan JW. Paraneoplastic retinopathies and optic neuropathies. Surv Ophthal 2003;48:12-38.
3. Campo E, Brunier MN, Merino MJ. Small cell cacinoma of the endometrium with associate ocular paraneoplastic syndrome. Cancer 1992;69:2283-8.
4. Cogan DG, Kuwabara T, Currie J, Kattah J. Paraneoplastic retinopathy simulating cone dystrophy with achromatopsia. Klin Monatsbl Augenheilkd 1990;197:156-8.
5. Jacobson DM, Thirkill CE. Paraneoplastic cone dysfunction: An usual visual remote effect of cancer. Arch Ophthalmol 1995;113:1580-2.
6. Milam AH. Clinical aspects: Paraneoplastic retinopathy, in Djamgoz MBA, Archer SN, Vellerga S (eds). Neurobiology and Clinical Aspects of the Outer Retina. London, Chapman and Hall 1995;461-71.
7. Khan N, Huang JJ, Foster SC. Cancer associated retinopathy (CAR): An autoimmune-mediated paraneoplastic syndrome. Semin Ophthalmol 2006;21:135-41.
8. Ling CP, Pavesio C. Paraneoplastic syndromes associated with visual loss. Curr Opin Ophthalmol 2003;14:426-32.
9. Adamus G, Aptsiauri N, Guy J, et al. Anti-enolase antibodies in cancer associated retinopathy [abstract]. Invest Ophthalmol Vis Sci 1993;34(suppl):s1485.
10. Keltner JL, Thirkill CE, Yip PT. Clinical and immunologic characteristics of melanoma-associated retinopathy syndrome: Eleven new cases and a review of 51 previously published cases. J Neuroophthalmology 2001;21:173-87.
11. Lu Y, Jia L, He S, Hurley MC, et al. Melanoma-associated retinopathy: A paraneoplastic autoimmune complication. Arch Ophthalmol 2009;127:1572-80.
12. Lei B, Bush RA, Milam AH, Sieving PA. Human melanoma-associated retinopathy (MAR) antibodies alter the retinal ON-response of the monkey ERG in vivo. Invest Ophthalmol Vis Sci 2000;41;262-6.
13. Wolf JE, Arden GB. Selective magnocellular damage in melanoma-associated retinopathy: Comparison with congenital stationary night blindness. Vision Res 1996;36:2369-79.
14. Nieuwendijk TJ, Hooymans JM. Paraneoplastic vitelliform retinopathy associated with metastatic choroidal melanoma. Eye (Lond) 2007;21:1436-7.
15. Zacks DN, Pinnolis MK, Berson EL, Gragoudas ES. Melanoma-associated retinopathy and recurrent exudative retinal detachments in a patient with choroidal melanoma. Am J Ophthalmol;132:578-81.
16. Jampol LM, Kim HH, Bryar PJ, Shankle JB, et al. Multiple serous retinal detachments and subretinal deposits as the presenting signs of metastatic melanoma. Retina 2004;24:320-2.
17. Sotodeh M, Paridaens D, Keunen J, van Schooneveld M, et al. Paraneoplastic vitelliform retinopathy associated with cutaneous or uveal melanoma and metastases. Klin Monbl Augenheilkd 2005;222:910-4.
18. Borkowski LM, Grover S, Fishman GA, Jampol LM. Retinal findings in melanoma-associated retinopathy. Am J Ophthalmol 2001;132:273-5.
19. Palmowski AM, Haus AH, Pföhler C, Reinhold U, et al. Bilateral multifocal chorioretinopathy in a woman with cutaneous malignant melanoma. Arch Ophthalmol 2002;120:1756-61.
20. Koreen L, He SX, Johnson MW, Hackel RE, et al. Anti-retinal pigment epithelium antibodies in acute exudative polymorphous vitelliform maculopathy: A new hypothesis about disease pathogenesis. Arch Ophthalmol 2011;129:23-9.
21. Eksandh L, Adamus G, Mosgrove L, Andréasson S. Autoantibodies against bestrophin in a patient with vitelliform paraneoplastic retinopathy and a metastatic choroidal malignant melanoma. Arch Ophthalmol 2008;126:432-5.
22. Case of the month: Melanoma associated retinopathy. West Coast Retina. 2011; from
http://westcoastretina.com/WestCoastRetina/Jan-2011.html.
23. Gass JDM, Chuang EL, Granek H. Acute exudative polymorphous vitelliform maculopathy. Trans Am Ophthalmol Soc 1988;86:354-66.
24. Gass JDM. Acute idiopathic exudative polymorphous vitelliform maculopathy. In: Gass JDM, ed. Stereoscopic atlas of macular diseases: Diagnosis and treatment, 4th ed. St Louis: CV Mosby, 1997:168-9, vol 1, 696-7, vol 2.
25. Chan CK, Gass JD, Lin SG. Acute exudative polymorphous vitelliform maculopathy syndrome. Retina 2003;23:453-62.
26. Machemer R. Zur Pathogenese des flachenhaften malignen Melanoms. Klin Monatsbl Augenheilkd 1966;148:641-52.
27. Gass JD, Gieser RG, Wilkinson CP, Beahm DE, Pautler SE. Bilateral diffuse uveal melanocytic proliferation in patients with occult carcinoma. Arch Ophthalmol 1990;108:527-33.
28. O’Neal KD, Butnor KJ, Perkinson KR, Proia AD. Bilateral diffuse uveal melanocytic proliferation associated with pancreatic carcinoma: A case report and literature review of this paraneoplastic syndrome. Surv Ophthalmol 2003;48:613-25.
29. Chahud F, Young RH, Remulla JF, Khadem JJ, Dryja TP. Bilateral diffuse uveal melanocytic proliferation associated with extraocular cancers: Review of a process particularly associated with gynecologic cancers. Am J Surg Pathol 2001;25:212-8.
30. Gass JDM, Glatzer RJ. Acquired pigmentation simulating Peutz-Jeghers syndrome: Initial manifestation of diffuse uveal melanocytic proliferation. Br J Ophthalmol 1991;75:693-5.
31. Heckenlively JR, Ferreyra HA. Autoimmune retinopathy: A review and summary. Semin Immunopathol 2008;30:127-34.
32. Monson DM, Smith JR. Acute zonal occult outer retinopathy. Surv Ophthalmol 2011;56:23-35.
33. Gass JDM. Acute zonal occult outer retinopathy. Donders Lecture: The Netherlands Ophthalmological Society, Maastricht, Holland, June 19, 1992. J Clin Neuroophthalmol 1993;13:79-97.
34. Francis PJ, Marinescu A, Fitzke FW, Bird AC, Holder GE. Acute zonal occult outer retinopathy: Towards a set of diagnostic criteria. Br J Ophthalmol 2005;89:70-3.
35. Bazhin AV, Shifrina ON, Savchenko MS, et al. Low titre autoantibodies against recoverin in sera of patients with small cell lung cancer but without a loss of vision. Lung Cancer 2001;34:99–104.
36. Savchenko MS, Bazhin AV, Shifrina ON, et al. Antirecoverin autoantibodies in the patient with non-small cell lung cancer but without cancer-associated retinopathy. Lung Cancer 2003;41:363–7.
37. Shimazaki K, Jirawuthiworavong GV, Heckenlively JR, Gordon LK. Frequency of anti-retinal antibodies in normal human serum. J Neuroophthalmol 2008;28:5-11.
38. Yannuzzi LA. The Retinal Atlas. Philadelphia, Elsevier 2010;689-690.
39. Reddy S, Finger PT. Unilateral diffuse uveal melanocytic proliferation (DUMP). Br J Ophthalmol 2007;91:1726-7.
40. Jampol LM, Fishman GA. Immunosuppression for autoimmune retinopathy. Arch Ophthalmol 2009;127:573-575.
41. Heckenlively JR, Jordan BL, Aptsiauri N. Association of antiretinal antibodies and cystoid macular edema in patients with retinitis pigmentosa. Am J Ophthalmol 1999;127:565-73.
42. Heckenlively JR, Fawzi AA, Oversier J, Jordan BL, Aptsiauri N. Autoimmune retinopathy: Patients with antirecoverin immunoreactivity and panretinal degeneration. Arch Ophthalmol 2000;118:1525-33.