Presentation
A 29-year-old Middle-Eastern female was referred to Wills Eye Hospital for evaluation of swelling of the left medial canthal region that had gradually enlarged over the course of a year. The patient denied having pain, discomfort, blurry vision, diplopia, pain with eye movements, erythema, epiphora, drainage, pulsatility, skin lesions, fevers, chills, malaise or fatigue. There was no history of trauma.
Medical History
 |
|
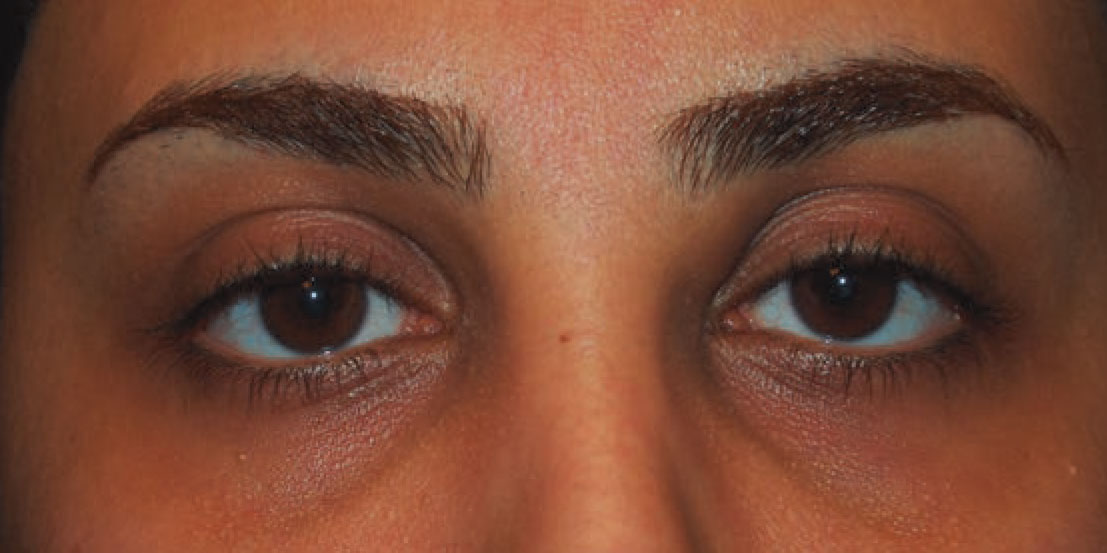
Figure 1. External photograph depicting swelling of the left medial canthal region that gradually enlarged over one year. |
Past medical history was notable only for hypothyroidism controlled with levothyroxine 75 mcg daily. Her surgical history included a cosmetic rhinoplasty several years prior to presentation. Family history was negative for chronic conditions. She denied alcohol, tobacco and illicit drug use.
Exam
The patient’s vital signs were stable and within normal limits. Visual acuity was 20/20 in both eyes. She had no relative afferent pupillary defect. Confrontational visual fields and extraocular movements were full bilaterally. She was orthophoric in primary gaze. Intraocular pressure was within normal limits bilaterally. External examination showed a palpable, mobile, subcutaneous mass of the left medial canthus (Figure 1, above). No proptosis was noted. Anterior slit lamp examination was unremarkable. She did not have palpable lymphadenopathy.
What's your diagnosis? What further workup would you pursue? The diagnosis appears below.
 |
Work-up, Diagnosis, and Treatment
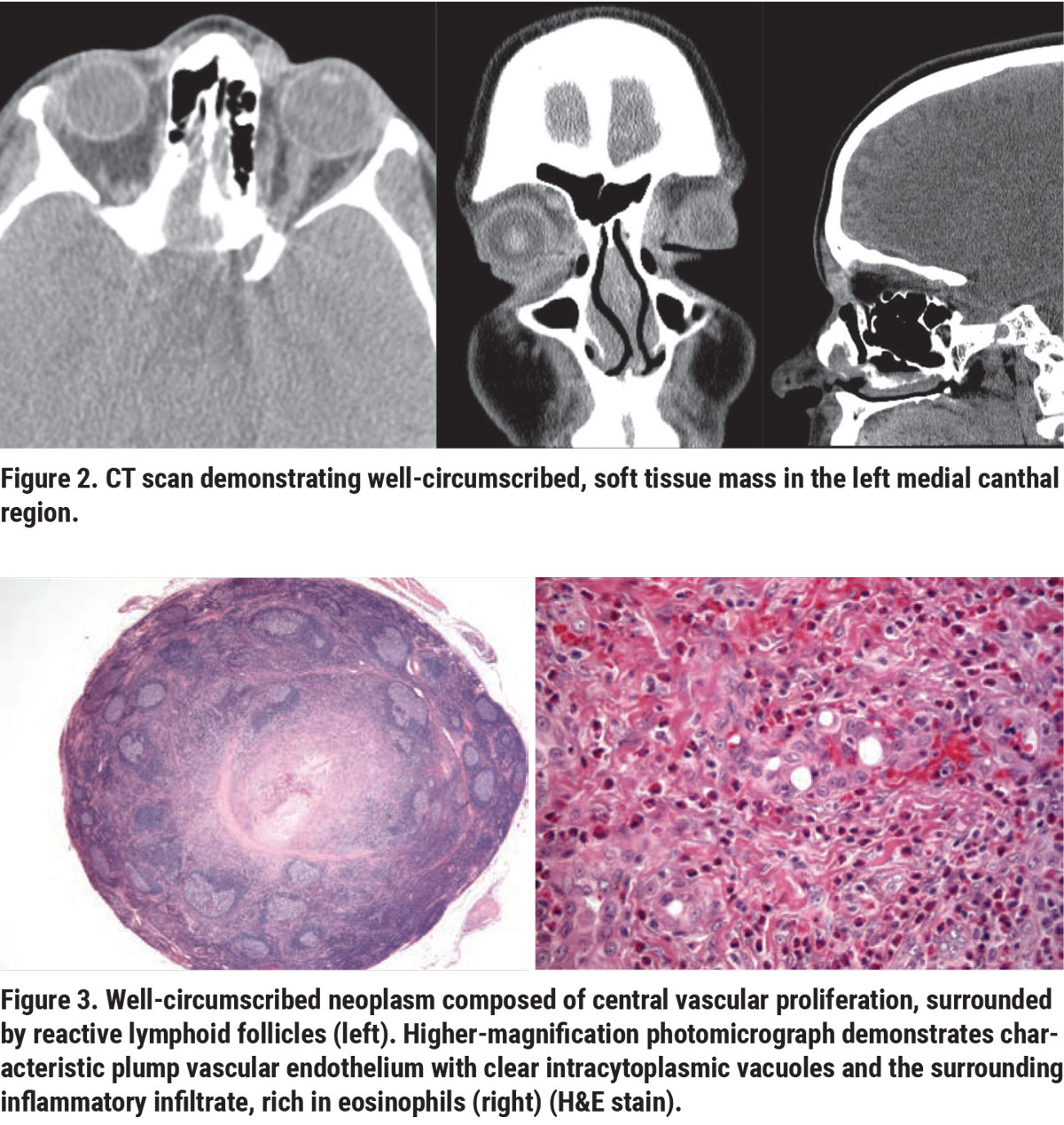
CT scan revealed a well-circumscribed, soft tissue mass in the left medial canthal region. There was no evidence of destruction or remodeling of the adjacent bone. The paranasal sinuses were clear (Figure 2, right). Based on clinical and radiographic features, the differential diagnosis for an indolent, painless, circumscribed, extraconal orbital mass in a young patient was broad. Entities considered included congenital lesions (dermoid cyst), fibrocystic lesions (solitary fibrous tumor), peripheral nerve sheath lesions (schwannoma, neurofibroma), vascular lesions (cavernous-venous malformation, arteriovenous malformation, lymphatic-venous malformation), secondary orbital lesions (mucocele), inflammatory lesions (IgG4-related sclerosing disease, sarcoidosis) and, less likely, foreign body due to remote trauma, infectious etiologies (orbital abscess, dacryocystitis), and malignant tumors (soft tissue tumors, lymphoma, metastasis).
The patient underwent left orbitotomy with excision of the mass. The lesion was well-circumscribed, tan and red in color, rubbery, and measured 16 x 9 x 8 mm. Histopathology showed a circumscribed proliferation of mostly small-caliber vascular channels associated with a larger vessel. The vascular channels were variably cellular and lined by epithelioid endothelium with focal tombstone configuration and occasional intracytoplasmic vacuoles. A prominent inflammatory response, rich in eosinophils, was associated with the vascular proliferation. The periphery of the lesion was rimmed by reactive follicles (Figure 3, above). No lymph node tissue was identified. There was no evidence of significant nuclear atypia, cellular sheeting, necrosis or mitotic activity. Immunohistochemical stains for CD31 and ERG highlighted the vascular endothelial proliferation, tombstoning and intracytoplasmic vacuoles. The histopathologic findings were compatible with epithelioid hemangioma. The postoperative period was uneventful, and no additional treatment was required. No recurrence of the lesion was noted at two-year follow-up.
Discussion
Epithelioid hemangioma (EH), also known as angiolymphoid hyperplasia with eosinophilia (ALHE) is an uncommon, benign vascular proliferative disorder.1,2 The condition was first described in 1969.1 EH most frequently occurs in the subcutaneous tissue or dermis of the head and neck region and can rarely affect the orbit and ocular adnexa.2,3 Previously reported ocular adnexal sites of EH include the eyelid, intraconal and extraconal soft tissue, lacrimal gland and conjunctiva.3-11 The patients with ocular adnexal EH most commonly present with symptoms that include swelling, proptosis, diplopia, ptosis, vision loss, pruritis, epiphora and/or pulsation.2-16 The pathogenesis of EH remains unclear at this time; proposed etiologies include neoplasm of endothelial cell origin, reactivity secondary to vascular injury, or a process secondary to an arteriovenous shunt.4,15 Malignant transformation has not been reported.
Histopathologic studies are essential for accurate diagnosis of EH, as the presenting clinical and radiologic signs are often nonspecific.3 The classic histopathological finding in EH is proliferation of “plump,” vacuolated, epithelioid-appearing endothelial cells that line vascular channels. Vascular proliferation of capillaries, arterioles and venules is accompanied by chronic inflammatory infiltrate, which may include lymphocytes, plasma cells, numerous eosinophils and lymphoid follicles.3,4,15 Immunohistochemically, vascular endothelial cells are typically positive for markers CD34, CD31, Factor VIII and ERG and may co-express lymphatic endothelial marker D2-40.15
EH must be distinguished from Kimura’s disease, which can closely resemble EH histopathologically, and in the past was regarded as an entity on a spectrum with EH.1,3 Kimura’s disease has a predilection toward young Asian males and is characterized by prominent lymphadenopathy, peripheral blood eosinophilia, elevated serum IgE and possible associated nephrotic syndrome. In the ocular adnexa, Kimura’s disease classically affects the lacrimal gland and lacks the characteristic vascular proliferation of EH.3,4,13 By contrast, EH tends to occur more frequently in females in their third or fourth decade of life, and is not associated with systemic manifestations like Kimura’s disease.2,4
EH also must be distinguished histopathologically from other vascular neoplasms of borderline malignancy (epithelioid hemangioendothelioma) and malignant vascular neoplasms (Kaposi sarcoma and angiosarcoma). Absence of significant nuclear atypia, cellularity and/or mitotic activity favors EH over more aggressive tumors. Additionally, EH lacks CAMTA1 and TFE3 gene rearrangements that may be observed in epithelioid hemangioendothelioma.15 Presence of reassuring features like FOSB gene rearrangement may support the diagnosis of EH.15
The traditional treatment of choice for EH is complete surgical excision. The recurrence rate may be as high as 33 percent, which is most likely attributable to lesions with poorly demarcated margins.4,13 Well-circumscribed lesions, such as that in our patient, tend to have low recurrence rates.14 In cases with extensive involvement, traditional treatment methods including systemic steroids, intralesional steroids, cryotherapy, radiotherapy, cyclophosphamide and subcutaneous methotrexate have been tried with limited success.4,12,14,18 In less-severe cases, observation for spontaneous regression following incisional biopsy and tissue diagnosis may be appropriate.2
More recently, systemic propranolol has been proposed as a treatment for EH, based upon its efficacy in regression of pediatric capillary hemangiomas.17 Proposed mechanisms of action include vasoconstriction, inhibition of angiogenesis and induction of apoptosis.16 One case of an epithelioid hemangioma with rapid progression, despite two subtotal excisions, steroids and chemotherapy agents, completely resolved with systemic propranolol.12 A second case of EH that couldn’t be fully excised due to adherence to surrounding tissues was also treated successfully with oral propranolol.16 Several case reports have described EH in which angiography identified high-level arterial flow, with subsequent embolization of vessels to decrease blood flow prior to surgical removal of the lesion.14.15 Finally, it’s been proposed that IL-5 inhibitors (e.g., mepolizumab) may be a promising treatment for EH, as seen in one patient with a refractory lesion and elevated IL-5 who experienced symptomatic relief with cytokine inhibition.18
In summary, we describe a patient with characteristic clinical, radiographic and histopathologic findings of EH and summarize the recent molecular genetics and management modalities for this unusual lesion. EH needs to be considered in the differential diagnosis of a circumscribed soft tissue vascular mass in a female patient. Complete excision is curative. Although this tumor can recur following incomplete excision, there have been no reports of malignant behavior.
1. Wells GC, Whimster IW. Subcutaneous angiolymphoid hyperplasia with eosinophilia. Br J Dermatol 1969;81:1:1-15.
2. Cunniffe G, Alonso T, Dinarès C, et al. Angiolymphoid hyperplasia with eosinophilia of the eyelid and orbit: The Western cousin of Kimura’s disease? Int Ophthalmol 2014;34:1:107.
3. Azari AA, Kanavi MR, Lucarelli M, et al. Angiolymphoid hyperplasia with eosinophilia of the orbit and ocular adnexa: Report of 5 cases. JAMA Ophthalmol 2014;132:5:633-636.
4. Mukherjee B, Kadaskar J, Priyadarshini O, et al. Angiolymphoid hyperplasia with eosinophilia of the orbit and adnexa. Ocul Oncol Pathol 2016;2:1:40-47.
5. Shields CL, Shields JA, Glass RM. Bilateral orbital involvement with angiolymphoid hyperplasia with eosinophilia (Kimura’s disease). Orbit 1990;9:2:89-95.
6. Hidayaat AA, Cameron JD, et al. Angiolymphoid hyperplasia with eosinophilia (Kimura’s disease) of the orbit and ocular adnexa. Am J Ophthalmol 1983;96:2:176-89
7. Sánchez-Acosta A, Moreno-Arredondo D, Rubio-Solornio RI, et al. Angiolymphoid hyperplasia with eosinophilia of the lacrimal gland: A case report. Orbit 2008;27:3:195-198.
8. Cook HT, Stafford ND. Angiolymphoid hyperplasia with eosinophilia involving the lacrimal gland: Case report. Br J Ophthalmol 1988;72:9:710-712.
9. Bostad L, Petterson W. Angiolymphoid hyperplasia with eosinophilia involving the orbita: A case report. Acta Ophthalmologica 1982;60:3:419-426.
10. Eisenberg E, Lowlicht R. Angiolymphoid hyperplasia with eosinophils: A clinical‐pathological conference. J Oral Maxillofac Pathol 1985;14:3:216-223.
11. Kennedy SM, Pitts JF, Lee WR, et al. Bilateral Kimura’s disease of the eyelids. Br J Ophthalmol 1992;76:12:755-757.
12. Moss HB, Sines DT, Blatt J, et al. Epithelioid hemangioma responsive to oral propranolol. Ophthalmic Plast Reconstr Surg 2012;28:4:e88-e90.
13. McEachren TM, Brownstein S, Jordan DR, et al. Epithelioid hemangioma of the orbit. Ophthalmology 2000;107:4:806-810.
14. Sedran L, Bonaso M, Mettus A, et al. Epithelioid hemangioma of the face. J Craniofac Surg 2018;29:8:e736-e739.
15. Stiefel HC, Ng JD, Wilson DJ, et al. Orbital cellular epithelioid hemangioma. Ocul Oncol Pathol 2019;5:6:424-431.
16. Lee SM, Khwarg SI, Ahn JH. Epithelioid hemangioma of the orbit treated with oral propranolol in adults. Can J Ophthalmol 2018;53:6:e244-e246.
17. Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med 2015;372:735-746.
18. Braun‐Falco M, Fischer S, Plötz SG, et al. Angiolymphoid hyperplasia with eosinophilia treated with anti‐interleukin‐5 antibody (mepolizumab). Br J Dermatol 2004;151:5:1103-1104.


