Inherited retinal diseases resulting from genetic mutations lead to progressive retinal degeneration and loss of visual function. Until recently, clinical characterization was the mainstay of diagnosis and classification of IRDs, but genetic characterization has been gaining increasing importance. Over the past three to four decades, more than 260 genes associated with IRDs have been identified, and another 37 mapped to chromosomes.1 This progress has led the way to the development of gene therapies to address disorders which were previously considered incurable.
This article will provide an overview of the principles of gene therapy and an update on the current active trials for the treatment of IRDs at the clinical stage of development.
Gene Therapy: Major Concepts
Genetic therapies address DNA mutations in several ways. First, the gene can be “augmented” by delivering correct copies of the genes to the affected cells, which will lead to synthesis of functional proteins. Gene augmentation is the most commonly used approach to IRDs. This approach targets autosomal recessive or X-linked mutations well, since blocked protein synthesis or production of an abnormal functionally null protein can be augmented and rescued by this technique.2
Second, the gene can be edited by “genome surgery” through delivery of “molecular scissors” called endonucleases to target cells, also known as Clustered Regularly Interspaced Short Palindromic Repeats” (CRISPR) technology. Lastly, the mRNA transcribed from the mutated DNA gene can be corrected or its translation stopped by the delivery of a binding anti-sense RNA segment, called “antisense oligonucleotide” (AON).
Except for AONs, the therapeutic genetic material must be delivered to target retinal cells using a vector. The most commonly used vectors are adeno-associated viruses (AAVs) and lentiviruses. Both are non-pathogenic and don’t cause serious systemic adverse effects. AAVs infect both dividing and non-dividing cells, do not integrate into the host genome and have a carrying capacity of 4.5 to 4.9 kb, a size corresponding to that of smaller genes.2 Lentiviruses have less propensity to infect non-dividing cells, integrate into the host genome with a consequent small risk of new oncogenic or non-oncogenic mutations, and can carry larger genes of up to 8 kb.2
After integrating the genetic material into vectors, the preparation is amplified, purified of empty capsids and supplemented with surfactant material to prevent product adherence to its container.3 The final product can be delivered via subretinal or intravitreal injection. The choice of the administration route depends mainly on the location of the cells that are targeted by the treatment.
Subretinal injection is preferred when outer retinal layers, including photoreceptors and retinal pigment epithelium, are affected, as is the case in most IRDs. Since delivery with this technique is localized, there’s minimal risk of extraocular dissemination and systemic immunogenicity. However, it requires pars plana vitrectomy under retrobulbar or general anesthesia, and can be subject to all vitrectomy-associated risks.3 As may be expected, subretinal injection induces a localized retinal detachment and/or thinning; however, this is usually transient and clinically non-significant.2-5
Intravitreal injection is chosen when internal retinal layers or wide areas of the retina must be treated, or when there are concerns of retinal fragility due to the underlying disease process that would prevent adequate and safe subretinal injection.3 Intravitreal administration is technically easier, but is associated with a higher risk of systemic shedding of the vector.3
 |
Targeting IRDs
IRDs are ideal targets for gene therapy for several reasons:
• the relative immune privilege of the eye;
• the tight blood-retinal barrier limiting systemic dissemination of substances injected intraocularly;
• the compartmentalized structure and small size of the eye, requiring injection of only small quantities of drugs for efficacy;
• the accessibility of the eye, allowing precise retinal structure visualization, targeted localized drug delivery and non-invasive monitoring of the patient’s response to therapy;
• the arrest of retinal cell proliferation after birth, resulting in indefinite expression of delivered genes after a single injection; and
• symmetric disease involvement of the contralateral eye that can be used as a control for evaluating a gene therapy’s efficacy.2
Among IRDs, those most amenable to genetic therapies are monogenic and caused by autosomal-recessive or X-linked mutations. Dominant mutations are more difficult to address, due to the gain-of-function abnormal protein interfering with the action of the correct protein that’s synthesized following treatment.2 Furthermore, slowly progressive IRDs diagnosed early in the disease process are more favorable candidates, since they allow for a wider window for treatment before the damage is too advanced and no viable target cells for gene therapy are left.3
Clinical Trials
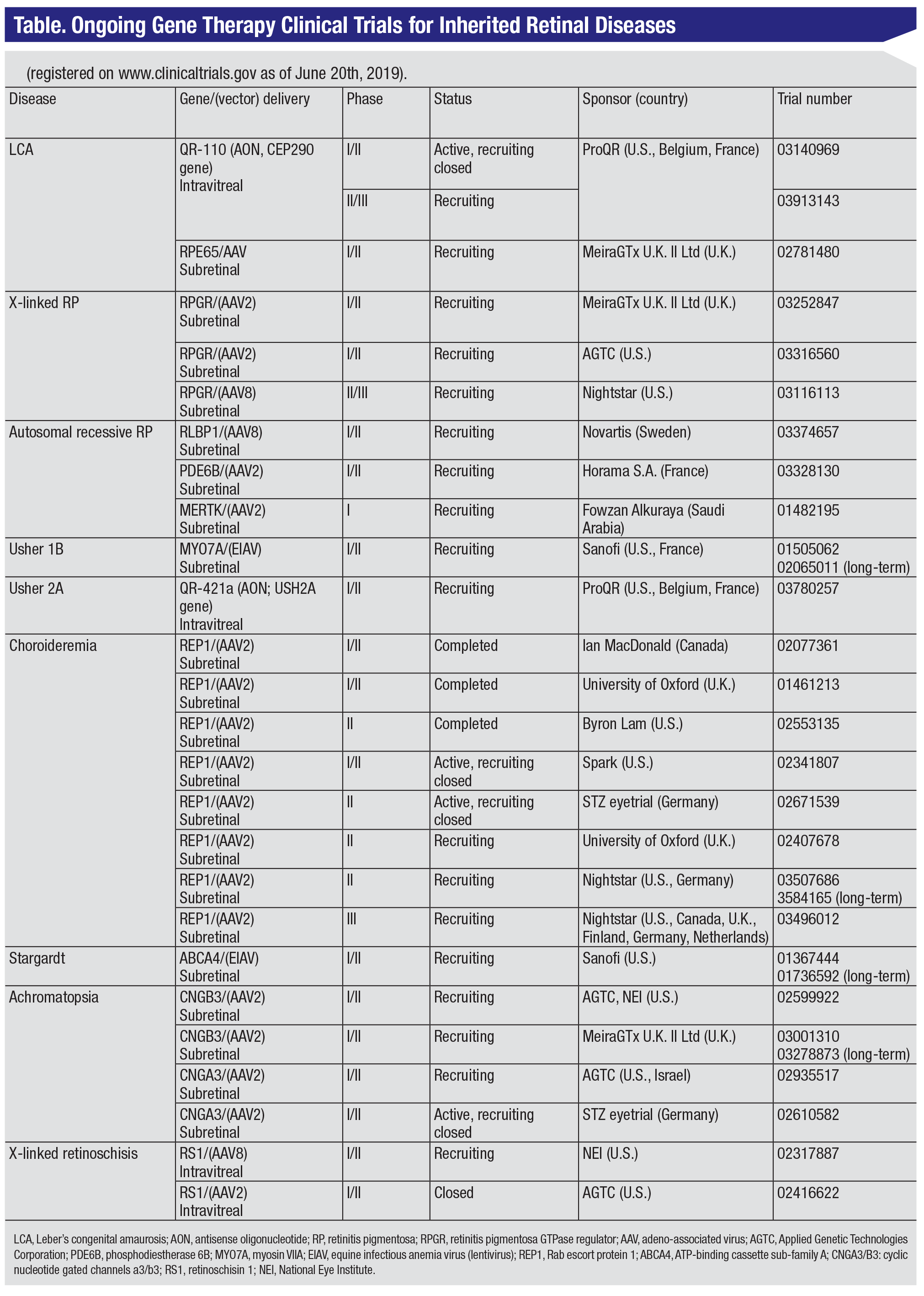
The expanding knowledge of the natural history of IRDs, the identification of significant visual function parameters, the development of non-invasive measurement instruments capable of detecting change in these parameters, and extensive preclinical research on animal models have enabled several gene therapies to reach the clinical stage of development (See Table, pg. 51).
• Leber congenital amaurosis (LCA). A landmark clinical trial led to the approval of voretigene neparvovec (Luxturna; Spark Therapeutics) by the U.S. Food and Drug Administration in December 2017, making it the only approved IRD genetic therapy.
Voretigene neparvovec is the most clinically advanced among studied gene therapies. It targets retinal pigment epithelial specific protein 65 kDa (RPE65)-related dystrophies, such as LCA type 2, and 1 to 3 percent of retinitis pigmentosa cases. The RPE65 protein is responsible for converting all-trans retinoid to 11-cis retinal, and its absence or dysfunction leads to an inability to regenerate visual pigment in photoreceptors. Voretigene neparvovec delivers correct copies of the RPE65 gene using an AAV vector. It’s administered as a single subretinal injection.
The Phase III trial studied 20 patients treated sequentially in both eyes with the drug, and nine control patients.6 At one year, treated patients met the primary endpoint of a significant improvement in performance in the multi-luminance mobility test when compared to control patients. Treated patients gained functional vision, allowing independent navigation over a wide range of luminance conditions, while subjects in the control group remained stable. Non-treated patients that were crossed over to the treatment group after one year achieved final outcomes similar to those in the original intervention group. No harmful immune responses associated with the drug itself were observed.
More than 400 other mutations across the 14 LCA-associated genes are known,2 and efforts to develop therapies targeting these other mutations continue. One of the most frequent LCA-associated mutations affects the centrosomal protein 290 (CEP290) gene, accounting for 15 percent of cases.2 The gene encodes a protein crucial for photoreceptor cilia function. Significant progress has been made in developing an AON, QR-110 (sepofarsen), aimed at correcting the CEP290-derived mRNA before its translation into protein. In Phase I/II trials, the drug was found to be safe and well-tolerated, as well as effective in terms of visual acuity improvement and its scores in multi-luminance mobility and full-field stimulus tests.7 A Phase II/III commercial clinical trial (ILLUMINATE) sponsored by ProQR evaluating QR-110 is currently ongoing.7
• Retinitis pigmentosa. The most common IRD is RP.8 It’s a genetically and phenotypically diverse disorder, characterized by primary rod and secondary cone photoreceptor degeneration.2 More than 100 associated mutated genes have been identified,1 and they collectively affect between 1 in 4,000 to 1 in 2,500 individuals.2 The disease can be inherited in an autosomal recessive (50 to 60 percent), autosomal dominant (30 to 40 percent) or X-linked fashion (5 to 15 percent).2 AAV-based gene augmentation therapies are underway for three autosomal recessive forms, currently at the Phase I/II stage of clinical development (Table, pg. 51). One of the more prevalent of these forms is a mutation in the phosphodiesterase 6B (PDE6B) gene, accounting for 2 to 4 percent of RP cases.9 The encoded PDE6B protein is a subunit of PDE6, that plays an essential role in the rod phototransduction cascade. Retinaldehyde-binding protein 1(RLBP1) gene mutations are much rarer9 and lead to abnormal rod retinoid metabolism. Diagnosis of the RLBP1-associated autosomal recessive form is made early due to specific fundus findings, leaving a wide therapeutic window for gene treatment.
Mer receptor tyrosine kinase (MERTK) gene mutations are also relatively rare.9 The MERTK protein is involved in outer photoreceptor segment phagocytosis by the RPE; these segments accumulate and lead to outer retinal degeneration when the protein is dysfunctional. Early findings are available for the MERTK gene therapy trial: Of the six patients included, none had any drug-related complications, and three experienced an improvement in visual acuity, although this wasn’t maintained at two years.10 X-linked RP cases are at the more severe end of the spectrum of the disease, and 70 to 80 percent are caused by mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene. Three independent commercial clinical trials—two Phase I/II trials and one Phase II/III trial—evaluating AAV-based RPGR gene therapy are currently underway.
• Usher syndrome. In Usher syndrome, RP is associated with audio-vestibular impairment; the disease accounts for approximately 20 to 40 percent of recessive RP.9 Genetically heterogeneous, it’s associated with 11 loci and nine genes important to the function and stability of the photoreceptor cilium. Clinical classification of Usher syndrome (I, II or III) is based on the severity and timing of the sensory impairments.2 The most severe type is Usher 1B, which is caused by a mutation in the myosin VIIa (MYO7A) gene.2 A commercial clinical trial using an equine lentivirus to deliver the large MYO7A gene subretinally, which includes a dose-escalation phase and a long-term follow-up phase, is currently ongoing. Early findings from four patients were presented in 2015: Besides the foveal detachment and transitory decrease in vision induced by the subretinal injection, there were no drug-related complications and no sustained surgical sequellae.5
Usher 2A is a less severe form of the syndrome. The causative mutation is found in the USH2A gene, which encodes usherin—an intercellular adhesion protein located at the basal aspect of the photoreceptor cilia. The STELLAR Phase I/II trial conducted by ProQR Therapeutics aims to evaluate the safety and efficacy of QR-421a, an AON administered intravitreally to correct the USH2A-derived mRNA. Three-month interim analysis results should be released in early 2020.11
• Choroideremia. This is an X-linked genetic disorder affecting the outer retina and choroid, characterized by progressive peripheral visual field loss, nyctalopia and total blindness within the first 30 years of life. It’s caused by a mutation in the choroideremia (CHM) gene encoding the Rab escort protein 1 (REP1), which is crucial to photoreceptor intracellular trafficking processes. Several groups have been working on treatments targeting this mutation. Three Phase I/II trials including a total of 26 patients at different stages of the disease, evaluating subretinal injections of AAV-based CHM gene therapies, are already completed. Results suggest relative safety with no severe systemic adverse effects,4,12,13 and with only two cases of localized retinal inflammation related to vector delivery.4,13 Formation of macular atrophic holes or severe retinal thinning are of concern in patients with choroideremia, whose degenerating retina is already thin and can significantly stretch during drug injection. This complication has been described in three treated cases.4,13 Optimization of the surgical technique to incorporate intraoperative OCT, which can identify the correct injection plane and rapidly detect retinal thinning, is expected to circumvent this complication.13 Effectiveness results in terms of visual acuity are also promising, with a significant improvement of 4.5 letters in treated eyes compared to a loss of 1.5 letters on average in control eyes. Results were sustained at two years of follow-up.13,14 Based on its encouraging Phase I/II findings, the company Nightstar Therapeutics has initiated a Phase III clinical trial.15
• Stargardt disease. Stargardt’s is an inherited macular degeneration. The most common causative mutation affects the ATP-binding cassette sub-family A (ABCA4) gene. The dysfunctional protein normally transports potentially toxic bisretinoid compounds from photoreceptors to the RPE.2 The ABCA4 gene is relatively large, requiring the use of an equine lentiviral vector for gene therapy development. A Phase I/II trial is presently investigating the safety and efficacy of its subretinal delivery.
Preliminary results following completion of the dose-escalation cohorts (Phase I) including a total of 15 patients, suggest that the drug is relatively safe.16 Notable adverse effects related to surgery and/or treatment included prolonged, but mild, IOP elevation in one case, subretinal fluid along the superior vascular arcade in another case, lymphopenia in another patient and complaints of “flashes and floaters” in several subjects.16 Phase II and long-term follow-up of Phase I patients are ongoing.
• Achromatopsia. A rare progressive cone degeneration beginning in early childhood, achromatopsia is characterized by poor visual acuity, hemeralopia and complete colour blindness. More than 75 percent of cases are caused by mutations in genes encoding cone-specification channels, namely cyclic nucleotide gated channel α3 and β3 (CNGA3 and CNGB3, respectively).2
Several independent Phase I/II trials relying on subretinally-injected AAV vectors containing correct copies of one of these two genes have been initiated, but results are not yet available.
• X-linked retinoschisis. This condition causes a localized splitting of the retina in the macular area. The usual onset is early childhood, and it afflicts 1 in 25,000 to 1 in 5,000 males. The protein encoded by the mutated retinoschisin 1 (RS1) gene is likely involved in cell adhesion. AAV-based therapies supplementing this gene are currently at the Phase I/II development stage.
Concerns about the weakness of the retina and the possibility of damage from subretinal injections, as well as involvement of inner retinal layers, have justified the use of an intravitreal approach. An AGTC-sponsored Phase I/II trial, including a total of 27 X-linked retinoschisis patients, has shown general safety and tolerability of the AAV-based drug, with only mild transitory ocular inflammation. However, no clinical effectiveness over the six-month interim analysis period was seen.17 These results have led to AGTC’s decision to continue monitoring of enrolled patients as per the study protocol, but not to further develop the product.17
Initial findings of the National Eye Institute’s Phase I/II trial investigating intravitreal treatment with another RS1-gene product in nine patients have been more promising, confirming systemic safety and suggesting efficacy, with retinal cavities transiently closing in one patient.18 The trial continues, and additional doses are being explored to pursue evidence of efficacy.18
Genetic Testing
Given these advancements in genetic therapeutic approaches for IRDs, genetic testing of afflicted patients to identify those who could benefit from these treatments is now more relevant than ever. Furthermore, a genetic diagnosis is of importance for family planning, financial decisions and career orientation. Genotype diagnosis is now part of the official guidelines directing the assessment of IRD patients.19
Sensitive and specific genetic tests are available for multiple IRDs, and a mutation is identified in 60 to 80 percent of those tested.20 Several types of tests can be ordered, including single-gene tests; gene panels, grouped by clinical diagnosis; whole exome sequencing, looking for mutations in the coding DNA; and whole genome sequencing, searching for mutations in both coding and non-coding DNA.3 The wider the search, the higher the chance of discovering IRD-causing mutations, but also other unrelated mutations or variants of unknown significance. Thus, the most specific test possible should be requested to avoid confusing results, as well as financial and emotional costs to the patient in case of unrelated abnormal findings.21 The patient should be offered professional genetic counseling both before and after testing.21 Most ophthalmologists aren’t trained to choose among available genetic tests, interpret their results and counsel patients as to their significance, so it’s paramount that an IRD specialist, a medical geneticist and/or a genetic counselor be involved.
In summary, Luxturna is the first FDA-approved gene therapy; it targets RPE65-related LCA. Other genetic therapies for IRDs are already on the way, with 23 of them in Phase I, II or III clinical trials. Most of them are gene-augmentation therapies that deliver correct copies of the gene to the retina; however, two AON-based therapies, acting at the level of the incorrect mRNA transcription, are also being studied. Three gene therapies are currently in more advanced Phase III trials, namely an RPGR-gene drug for X-linked RP, an REP1-gene therapy for choroideremia and an AON—QR-110—for the treatment of LCA. Many more potential therapies are still in preclinical development.2
Although gene therapies for IRDs have come a long way, overcoming many barriers to clinical development, challenges remain related to choice and optimization of drug delivery techniques and selection of the optimal timing for treatment. With other upcoming therapeutic options for patients who have advanced stages of IRDs, such as optogenetics and stem cell therapies, exciting times lie ahead. REVIEW
Dr. Bostan is co-chief resident in ophthalmology, and Dr. Qian is an assistant professor of Adult and Pediatric Vitreoretinal Surgery and Diseases, at the Centre Universitaire d’Ophtalmologie (CUO) at the University of Montreal, Maisonneuve-Rosemont Hospital in Montreal and the Centre Hospitalier Universitaire (CHU) Sainte-Justine in Montreal. Dr. Qian is also the Director of the Inherited Retinal Diseases service and the Electrophysiology Laboratories at the University of Montreal. Dr. Bostan and Dr. Qian have no financial interest in any product mentioned.
1. RetNet. Summaries of genes and loci causing retinal disease. Retinal Information Network 2018. https://sph.uth.edu/retnet/sum-dis.htm. Accessed 20 June 2019.
2. Lipinski DM, Thake M, MacLaren RE. Clinical applications of retinal gene therapy. Progress in Retinal and Eye Research 2013;32:22-47.
3. Leroy BP, Pennesi ME, Ohnsman CM. Brave New World: Gene Therapy for Inherited Retinal Disease. Supplement (CME Accredited Activity). Accessed 1 July 2018
4. Lam BL, Davis JL, Gregori NZ, MacLaren RE, Girach A, Verriotto JD, et al. Choroideremia gene therapy phase 2 clinical trial: 24-Month results. American Journal of Ophthalmology 2019;197:65-73.
5. Weleber R, Stout T, Lauer A, Pennesi M, Audo I, Mohand-Said S, et al. Early findings in a Phase I/IIa clinical program for Usher syndrome 1B (USH1B; MIM #276900). Investigative Ophthalmology & Visual Science 2015;56:7:2286.
6. Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017;390:10097:849-60.
7. ProQR news release. ProQR announces positive interim results from phase 1/2 clinical trial of QR-110 in LCA10 patients, and plans to start a phase 2/3 pivotal trial: ProQR 2018. https://ir.proqr.com/news-releases/news-release-details/proqr-announces-positive-interim-results-phase-12-clinical-trial. Accessed 20 June 2019.
8. Bocquet B, Lacroux A, Surget MO, Baudoin C, Marquette V, Manes G, et al. Relative frequencies of inherited retinal dystrophies and optic neuropathies in Southern France: Assessment of 21-year data management. Ophthalmic Epidemiology 2013;20:1:13-25.
9. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368:9549:1795-809.
10. Ghazi NG, Abboud EB, Nowilaty SR, Alkuraya H, Alhommadi A, Cai H, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: Results of a phase I trial. Human Genetics 2016;135:3:327-43.
11. ProQR news release. About Usher syndrome type 2: ProQR Therapeutics 2019. https://www.proqr.com/qr-421a-for-usher-syndrome-type-2/. Accessed 20 June 2019.
12. Dimopoulos IS, Hoang SC, Radziwon A, Binczyk NM, Seabra MC, MacLaren RE, et al. Two-year results after AAV2-mediated gene therapy for choroideremia: The Alberta experience. American Journal of Ophthalmology 2018;193:130-42.
13. Xue K, Jolly JK, Barnard AR, Rudenko A, Salvetti AP, Patricio MI, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nature Medicine 2018;24:10:1507-12.
14. Edwards TL, Jolly JK, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, et al. Visual acuity after retinal gene therapy for choroideremia. The New England Journal of Medicine 2016;374:20:1996-8.
15. Nightstar Therapeutics news release. Nightstar Therapeutics announces initiation of STAR phase 3 registrational trial for NSR-REP1 in choroideremia: First-ever phase 3 choroideremia gene therapy trial, 2018. https://www.globenewswire.com/news-release/2018/03/05/1414686/0/en/Nightstar-Therapeutics-Announces-Initiation-of-STAR-Phase-3-Registrational-Trial-for-NSR-REP1-in-Choroideremia.html. Accessed 21 June 2019.
16. Bakall B, Gamm D, Sohn EH, Gregori NZ, Duncan J, Michaelides M, et al. Retinal frontiers: Updates in gene therapy and stem cell therapy. Phase 1 gene therapy trials overview. (Academy Course). American Academy of Ophthalmology Annual Meeting; November 12, 2017; New Orleans.
17. AGTC news release. AGTC Announces Topline Interim Six-Month Data from Phase 1/2 X-Linked Retinoschisis Clinical Study; Termination of Biogen Collaboration: AGTC; 2018. http://ir.agtc.com/news-releases/news-release-details/agtc-announces-topline-interim-six-month-data-phase-12-x-linked. Accessed 20 June 2019.
18. Cukras C, Wiley HE, Jeffrey BG, Sen HN, Turriff A, Zeng Y, et al. Retinal AAV8-RS1 gene therapy for x-linked retinoschisis: Initial findings from a phase I/IIa trial by intravitreal delivery. Molecular Therapy 2018;26:9:2282-94.
19. American Academy of Ophthalmology. Clinical Statement: Recommendations on Clinical Assessment of Patients with Inherited Retinal Degenerations, 2016.
20. Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically focused molecular investigation of 1,000 consecutive families with inherited retinal disease. Ophthalmology 2017;124:9:1314-31.
21. Stone E, Aldave AJ, Drack AV, MacCumber MW, Sheffield VC, Traboulsi E, et al. Recommendations of the American Academy of Ophthalmology Task Force on Genetic Testing, 2014.


