Intraocular lymphomas are rare malignancies that display a wide array of clinical manifestations; therefore diagnosis can be challenging. Almost all intraocular lymphomas are non-Hodgkin’s lymphomas and the vast majority are of B-cell origin. Further confusion has arisen from the various nomenclature and classification schemes that have historically been used to describe this heterogeneous group of tumors. Many experts now group these entities based upon their site of ocular involvement and whether the disease is primary or secondary.
Using this scheme, the intraocular lymphomas can be subdivided into: primary vitreoretinal lymphoma; primary uveal lymphoma; and secondary intraocular manifestations of systemic lymphoma (See Table 1). Each form has distinct clinical features that help to establish the diagnosis and to guide treatment.
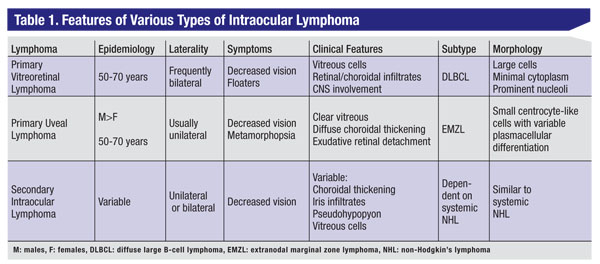 |
| ||||||||



Primary vitreoretinal lymphoma (PVRL) is a variant of primary central nervous system lymphoma (PCNSL), which is generally an aggressive, diffuse large B-cell malignancy associated with poor prognosis.1 The age-adjusted incidence of PCNSL in the Unites States is approximately 4.8 per million population, however, the exact incidence of PVRL is unknown.2 The association between PVRL and PCNSL is variable, with CNS disease manifesting prior to, following or occurring simultaneously with ocular presentation. Approximately 25 percent of patients with PCNSL will have concomitant PVRL.3 In contrast, 56 to 85 percent of individuals with PVRL ultimately develop central nervous system involvement.4-7 In immunocompetent individuals, the peak incidence of PVRL is between the fifth and seventh decade, although disease may occur at a younger age in the immunocompromised population.
Symptoms and Clinical Findings
Individuals may be asymptomatic, however the majority present with painless decrease in vision or floaters.6 Asymptomatic patients are often diagnosed when ophthalmic screening is performed in the setting of known PCNSL. The clinical findings are bilateral in 80 percent of cases, but are frequently asymmetric.4 The hallmark of PVRL is the presence of fine vitreous cells (See Figure 1) and subretinal pigment epithelium deposits (See Figure 2). When present, focal, multifocal or diffuse retinal, choroidal or chorioretinal infiltrates are considered essentially pathognomonic.8 Anterior segment findings can occur including keratic precipitates, iris nodules, aqueous cells and flare, but are non-specific.9 Other less-commonly reported features include: perivasculitis;6 retinal artery occlusion;11 exudative retinal detachment;12 multifocal “punched-out” lesions at the level of the RPE;13 and optic atrophy.14
Diagnostic Evaluation
Diagnostic evaluation should begin with a thorough history that explores not only ocular symptoms, but also changes in cognitive functioning, neurological decline and risk factors for immunocompromise. Due to the high correlation between PVRL and PCNSL, all patients diagnosed with ophthalmic disease should undergo systemic evaluation by an experienced oncologist. Recommended testing includes gadolinium-enhanced MRI of the brain and spinal cord, CSF studies, complete blood count and HIV testing, when appropriate.
In the setting of existing PCNSL, the diagnosis of PVRL with classic clinical findings is straightforward and biopsy of an ophthalmic site is unnecessary. In the absence of prior CNS disease, biopsy remains the gold standard. Several techniques exist including vitreous biopsy, retinal biopsy and subretinal biopsy. Proper surgical techniques and handling of the sample are critical, as aspirates are generally of low cellularity, and fragile lymphoma cells are prone to lysis during collection. For vitreous biopsy, an undiluted sample of approximately 1 to 2 mL is collected prior to the start of the infusion.15 Next, a second diluted vitreous specimen using gentle vitreous cutting is obtained.16 The vitreous cassette may be submitted as a third sample.17 Specimens should be delivered to the laboratory, without fixative, within one hour of surgery.15 PVRL cells are typically two to four times larger than normal lymphocytes, are pleomorphic and have scant cytoplasm.18 The nuclei may be round, oval or indented, with conspicuous nuclear membranes, occasional fingerlike protrusions, and multiple, prominent, eccentrically located nucleoli. Mitoses are frequently observed.19
| ||||


Differential Diagnosis
Delay in diagnosis of PVRL is common, as non-specific ophthalmic manifestations can masquerade as inflammatory, other neoplastic and infectious etiologies. In one series of 32 patients with histologically confirmed PVRL, the average interval between the onset of ocular symptoms and diagnosis was 21 months.7 The differential diagnosis includes: sarcoidosis; syphilis; tuberculosis; birdshot retinochoroidopathy; multifocal chorioretinitis; acute posterior multifocal placoid pigmentary epitheliopathy; serpiginous choroiditis; and punctate inner choroidopathy.22 When infiltrative subretinal or choroidal lesions are present, neoplasms such as choroidal metastases and amelanotic melanoma should be considered. In immunocompromised individuals, infectious diseases such as acute retinal necrosis, cytomegalovirus, toxoplasmosis and pneumocystis choroiditis should be considered.
Treatment
Consensus guidelines for treatment of PVRL have not been firmly established and depend upon patient factors as well as local expertise. When disease is unilateral and limited to the eye, local delivery of intravitreal methotrexate or rituximab has been shown to be effective and well-tolerated.23,24 The largest reported series treated with intravitreal methotrexate (400 mg in 0.1 ml of 0.9% sodium chloride) included 44 eyes in 26 patients using an induction-consolidation-maintenance regimen. Clinical remission was achieved in all cases following an average of 6.4 injections with 95 percent of eyes requiring 13 injections or less.25 None of the patients developed relapse after a follow-up period ranging from 41 to 107 months.25 The most commonly reported ocular side effects included cataract, conjunctival hyperemia and transient keratopathy.24 Early evidence suggests that rituximab may have fewer side effects and require fewer injections to achieve remission, however further investigation on larger numbers of patients is required.23 Large retrospective studies have shown that while ocular treatment appears to improve local control, there is no proven survival benefit of ocular therapy.26
Prior to acceptance of intravitreal chemotherapy, bilateral external beam radiotherapy was widely used. EBRT remains an important therapy, particularly in patients with bilateral involvement, for those who may not tolerate intravitreal chemotherapy, and in individuals who find it difficult to return for multiple injections. Irradiation doses have varied, with an average dose of approximately 40 Gy given in fractions of 1.5 to 2.0 Gy.27-30 Response to radiation and relapse rates are variable. In one series of 21 eyes in 12 patients, initial treatment included EBRT and systemic chemotherapy (six patients), systemic chemotherapy alone (four patients), EBRT alone (one patient), and no treatment (one patient). No ocular recurrence was observed in those receiving EBRT after a follow-up of four to 58 months (average of 18 months). Two patients who received chemotherapy but no EBRT developed ocular relapse at a follow-up time of 14 and 31 months.30 Radiation-related side effects included optic neuropathy, retinopathy, conjunctivitis, dry eyes, cataracts and glaucoma.31 In most cases, EBRT is reserved for individuals under age 65 years due to the high incidence of neurotoxicity in elderly individuals.
In patients with coexisting PCNSL, the majority are currently treated with high-dose intravenous chemotherapy. Methotrexate (8g/m2) is most commonly used, either as a single agent or as part of a combination regimen. In a large, multicenter, retrospective study of 221 immunocompetent patients with PCNSL with ocular involvement, the median progression-free survival and overall survival were 18 and 31 months, respectively.26
Primary Uveal Lymphoma
The primary uveal lymphomas can be further subdivided into choroidal, iridal and ciliary body lymphomas.
Primary choroidal lymphoma accounts for the majority of cases and is generally a low-grade B-cell lymphoma with a prolonged benign course. These tumors are morphologically and genetically similar to extranodal marginal zone lymphoma (EMZL) found elsewhere in the body. Primary iridal lymphomas may be of either B-cell or T-cell origin. The incidence of primarily iridal and ciliary body lymphoma is unknown, as these tumors have only been described in a handful of case reports. The discussion that follows pertains to primary choroidal lymphoma; while also rare, there are approximately 70 to 80 case reports in the literature.32
| ||||


Symptoms & Clinical Findings
Primary choroidal lymphoma is predominantly a unilateral process that most commonly affects men in the fifth to seventh decade. Initial symptoms may include recurrent episodes of painless, blurred vision and metamorphopsia secondary to exudative retinal detachment affecting the fovea.
When the disease is advanced, patients may develop pain and severely reduced vision due to secondary angle-closure glaucoma and extensive retinal detachment, respectively. When extraocular extension is present, proptosis and diplopia may occur. A classic feature is the presence of solitary or multiple yellow, creamy choroidal infiltrates (See Figure 3). Notably, the vitreous remains clear with the absence of cellular reaction. Ultimately, diffuse thickening of the uveal tract develops and is often associated with exudative retinal detachment. In some cases, there may be episcleral extension appearing as a non-mobile orange to yellow or “salmon” patch.
Diagnostic Evaluation
Several imaging techniques are useful for establishing the diagnosis. B-scan ultrasonography typically reveals choroidal thickening with low echogenicity. Both computed tomography and magnetic resonance imaging of the globes confirm choroidal thickening with a corresponding decrease in the size of the vitreous cavity. Calcification is absent. Fluorescein angiography demonstrates early hypofluoresence with multiple foci of hyperfluorescence and staining in the late phase. Biopsy of episcleral tumor nodules and choroidal aspirates may also aid in the diagnosis.
Histopathologically, the features of primary choroidal lymphoma are similar to EMZL located elsewhere in the body. The cells are generally of the centrocyte-like, monocytoid and plasmacytoid type. Dutcher bodies, or collections of intranuclear immunoglobulin, can often be observed. Immunohistochemistry confirms the expression of B-cell antigens (CD20 and CD79a). Gene rearrangement studies or flow cytometry support the clonality and neoplastic nature of these B-cells.
Differential Diagnosis
The differential diagnosis of primary choroidal lymphoma includes diffuse uveal melanoma, uveal effusion syndrome, posterior scleritis and uveal metastases. Diffuse uveal melanoma can usually be distinguished based upon its pigmented appearance, vascularity, disturbance of the RPE and faster growth rate.33 Uveal effusion syndrome is often bilateral and has associated dilated episcleral vessels, vitreous cells and “leopard spot-like” changes in the RPE. Fluorescein angiography does not generally demonstrate leakage.32 Posterior scleritis occurs more frequently in women, is associated with autoimmune disease and demonstrates high internal reflectivity with the classic “T-sign” on B-scan ultrasonography. In the majority of uveal metastases, a prior history of malignancy can be elicited. Additionally, B-scan ultrasonography usually demonstrates medium internal reflectivity and fluorescein angiography reveals widespread leakage in the late phase.
|

Before initiating localized treatment for primary choroidal lymphoma, it is essential to perform systemic studies to exclude the possibility of systemic lymphoma with uveal involvement. Recommended studies include CT of the chest and abdomen, complete blood count and serum protein electrophoresis.34 Once disease is confirmed to be limited to the choroid, management consists of low-dose EBRT administered over several fractions.34 Primary choroidal lymphoma typically follows a less-aggressive course than primary vitreoretinal lymphoma. In 13 eyes with biopsy proven primary choroidal lymphoma, subsequent systemic lymphoma developed in two cases (three and seven years after the initial diagnosis).35 This underscores the importance of staging at the time of diagnosis and periodic systemic surveillance.
Intraocular Manifestations of Systemic Lymphoma
Intraocular lymphoma secondary to disseminated systemic lymphoma most commonly affects the choroid. For this reason, secondary lymphoma can have a similar clinical appearance to primary choroidal lymphoma.36 Rarely, a spread to the retina (See Figure 4) without choroidal infiltration can occur.36 Other exceptional presentations of secondary intraocular lymphoma include pseudohypopyon and iris infiltration.37,38 While exceedingly rare, iridal lymphoma secondary to systemic non-Hodgkin’s lymphoma is more common than primary iridal lymphoma.32 The most common subtype of systemic lymphoma that affects the eye is diffuse large B-cell lymphoma. This is followed by multiple myeloma, extramedullary plasmacytoma, lymphoplasmocytic lymphoma/immunocytoma and marginal zone B-cell lymphoma (MALT lymphoma).36 The morphologic and immunophenotypic features of secondary choroidal lymphoma bear resemblance to its primary systemic counterpart. Staging evaluation and close collaboration with an experienced oncologist are critical in the management of patients with systemic lymphoma with intraocular involvement.
Drs Turell and Singh are at the Cole Eye Institute, Cleveland Clinic. Contact Dr. Singh at the Department of Ophthalmic Oncology, Cole Eye Institute, Cleveland Clinic, 9500 Euclid Ave., Cleveland, OH 44195. Phone: (216) 445 9479; fax: (216) 445 2226; e-mail:
singha@ccf.org.
Image credit, Figure 1. Reproduced with permission from Singh AD, Lewis H, Schachat AP. Primary lymphoma of the central nervous system. Ophthalmol Clin North Am 2005;18:199–207.
1. Pe’er J, Hochberg FH, Foster CS. Clinical review: Treatment of vitreoretinal lymphoma. Ocul Immunol Inflam 2009;17(5):299-306.
2. Olson JE, Janney CA, Rao RD, Cerhan JR, Kurtin PJ, Schiff D, et al. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer 2002;95(7):1504-10.
3. Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg 1988;68(6):835-53.
4. Peterson K, Gordon KB, Heinemann MH, DeAngelis LM. The clinical spectrum of ocular lymphoma. Cancer 1993;72(3):843-9.
5. Cassoux N, Merle-Beral H, Leblond V, Bodaghi B, Milea D, Gerber S, et al. Ocular and central nervous system lymphoma: clinical features and diagnosis. Ocul Immunol Inflam 2000;8(4):243-50.
6. Char DH, Ljung BM, Miller T, Phillips T. Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology 1988;95(5):625-30.
7. Freeman LN, Schachat AP, Knox DL, Michels RG, Green WR. Clinical features, laboratory investigations, and survival in ocular reticulum cell sarcoma. Ophthalmology 1987;94(12):1631-9.
8. Gass JD, Sever RJ, Grizzard WS, Clarkson JG, Blumenkranz M, Wind CA, et al. Multifocal pigment epithelial detachments by reticulum cell sarcoma. A characteristic funduscopic picture. Retina 1984;4(3):135-43.
9. Rajagopal R, Harbour JW. Diagnostic testing and treatment choices in primary vitreoretinal lymphoma. Retina 2011;31(3):435-40.
10. Fredrick DR, Char DH, Ljung BM, Brinton DA. Solitary intraocular lymphoma as an initial presentation of widespread disease. Arch Ophthalmol 1989;107:395-7.
11. Gass JD, Trattler HL. Retinal artery obstruction and atheromas associated with non-Hodgkin’s large cell lymphoma (reticulum cell sarcoma). Arch Ophthalmol 1991;109:1134-9.
12. Michelson JB, Michelson PE, Bordin GM, Chisari FV. Ocular reticulum cell sarcoma. Presentation as retinal detachment with demonstration of monoclonal immunoglobulin light chains on the vitreous cells. Arch Ophthalmol 1981;99:1409-11.
13. Lang GK, Surer JL, Green WR, Finkelstein D, Michels RG, Maumenee AE. Ocular reticulum cell sarcoma. Clinicopathologic correlation of a case with multifocal lesions. Retina 1985;5(2):79-86.
14. Purvin V, Van Dyk HJ. Primary reticulum cell sarcoma of the brain presenting as steroid-responsive optic neuropathy. J Clin Neuro-ophthalmol1984;4(1):15-23.
15. Coupland SE. Vitreous biopsy: Specimen preparation and interpretation. Monogr Clin Cytol. 2012;21:61-71. Epub 2011 Oct 18.
16. Margolis R, Brasil OF, Lowder CY, Singh RP, Kaiser PK, Smith SD, et al. Vitrectomy for the diagnosis and management of uveitis of unknown cause. Ophthalmology 2007;114:1893-7.
17. Yeh S, Weichel ED, Faia LJ, Albini TA, Wroblewski KK, Stetler-Stevenson M, et al. 25-Gauge transconjunctival sutureless vitrectomy for the diagnosis of intraocular lymphoma. Br J Ophthalmol 2010 May;94(5):633-8.
18. Farkas T, Harbour JW, Davila RM. Cytologic diagnosis of intraocular lymphoma in vitreous aspirates. Acta Cytol 2004 Jul-Aug;48(4):487-91.
19. Whitcup SM, de Smet MD, Rubin BI, Palestine AG, Martin DF, Burnier M, Jr., et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology 1993;100:1399-406.
20. Chan CC, Gonzales JA. Classification of lymphomas. Primary Intraocular Lymphoma. 1 ed. Hackensack, NJ: World Scientific Publishing Co. Pte. Ltd.; 2007.
21. Akpek EK, Maca SM, Christen WG, Foster CS. Elevated vitreous interleukin-10 level is not diagnostic of intraocular-central nervous system lymphoma. Ophthalmology 1999;106:2291-5.
22. Singh AD, Lewis, H, Schachat, AP, Peereboom D. Lymphoma of the retina and CNS. In: Singh A, Damato, BE, Pe’er J, Murphree AL, Perry, JD, editor. Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders Elsevier Inc.; 2007. p. 372-7.
23. Kitzmann AS, Pulido JS, Mohney BG, Baratz KH, Grube T, Marler RJ, et al. Intraocular use of rituximab. Eye (Lond). 2007;21(12):1524-7. Epub 2007/04/28.
24. Smith JR, Rosenbaum JT, Wilson DJ, Doolittle ND, Siegal T, Neuwelt EA, et al. Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvement. Ophthalmology 2002;109:1709-16.
25. Frenkel S, Hendler K, Siegal T, Shalom E, Pe’er J. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Brit J Ophthalmol 2008;92(3):383-8.
26. Grimm SA, McCannel CA, Omuro AM, Ferreri AJ, Blay JY, Neuwelt EA, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology 2008;71(17):1355-60.
27. Margolis L, Fraser R, Lichter A, Char DH. The role of radiation therapy in the management of ocular reticulum cell sarcoma. Cancer 1980;45(4):688-92.
28. Hormigo A, Abrey L, Heinemann MH, DeAngelis LM. Ocular presentation of primary central nervous system lymphoma: Diagnosis and treatment. British J Haematol 2004;126(2):202-8.
29. Isobe K, Ejima Y, Tokumaru S, Shikama N, Suzuki G, Takemoto M, et al. Treatment of primary intraocular lymphoma with radiation therapy: A multi-institutional survey in Japan. Leuk Lymphoma 2006;47(9):1800-5.
30. Berenbom A, Davila RM, Lin HS, Harbour JW. Treatment outcomes for primary intraocular lymphoma: Implications for external beam radiotherapy. Eye (Lond). 2007;21(9):1198-201.
31. Jahnke K, Thiel E, Abrey LE, Neuwelt EA, Korfel A. Diagnosis and management of primary intraocular lymphoma: An update. Clin Ophthalmol 2007;1(3):247-58.
32. Coupland SE. Uveal Lymphoproliferative Tumors. In: Singh AD, Damato B, Pe’er J, Murphree AL, Perry JD, editors. Clinical Ophthalmic Oncology. Philadelphia, PA: Elsevier, Inc; 2007. p. 316-21.
33. Font RL, Spaulding A, Zimmerman L. Diffuse malignant melanoma of the uveal tract: A clinicopathological report of 54 cases. Trans Am Acad Ophthalmol Otolaryngol. 1968;72:877-95.
34. Augsburger JJ, Greatrex KV. Intraocular lymphoma: Clinical presentations, differential diagnosis and treatment. Trans Pa Acad Ophthalmol Otolaryngol 1989;41:796-808. Epub 1989/01/01.
35. Coupland SE, Foss HD, Hidayat AA, Cockerham GC, Hummel M, Stein H. Extranodal marginal zone B cell lymphomas of the uvea: an analysis of 13 cases. Journal Pathol 2002;197(3):333-40.
36. Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Exp Ophthalmol 2008;36(6):564-78.
37. Shakin EP, Augsburger JJ, Eagle RC, Jr., Ehya H, Shields JA, Fischer D, et al. Multiple myeloma involving the iris. Arch Ophthalmol 1988;106:524-6.
38. Tranos PG, Andreou PS, Wickremasinghe SS, Brazier JD. Pseudohypopyon as a feature of multiple myeloma. Arch Ophthalmol 2002;120:87-8.


