A variety of pigmented choroidal lesions can be encountered in clinical practice, and differentiating between benign and malignant lesions is critically important for the ophthalmologist. Additionally, each individual lesion can have a heterogenous range of clinical presentations, often with overlap between different conditions that can lead to diagnostic uncertainty in select cases. Choroidal melanoma remains the most common concerning pigmented choroidal lesion. However, the following lesions can simulate melanoma:
- choroidal nevus;
- congenital hypertrophy of the retinal pigment epithelium;
- peripheral exudative hemorrhagic chorioretinopathy;
- choroidal melanocytosis;
- pigmented choroidal metastasis from cutaneous or choroidal melanoma;
- melanocytoma;
- bilateral diffuse uveal melanocytic proliferation;
- retinal pigment epithelium adenoma/adenocarcinoma; and
- suprachoroidal hemorrhage.
While a detailed clinical examination is the most important component in diagnosing these conditions, a thorough history and multimodal imaging can also be critical in correctly identifying the pigmented lesions listed above and choosing the appropriate management strategy.
Here, we’ll describe how we approach each of them.
Choroidal Melanoma
 |
|
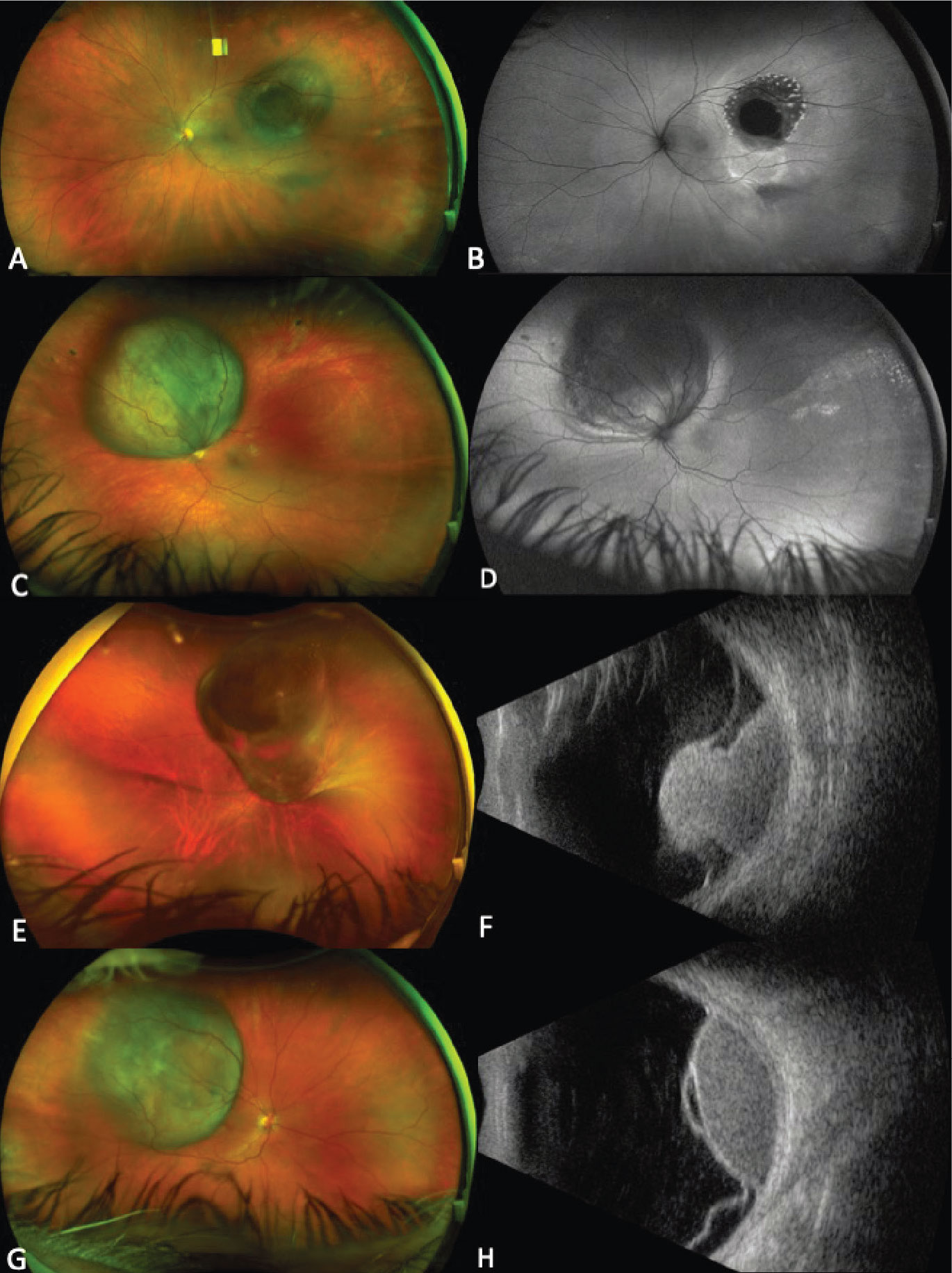
Figure 1. Ultra-widefield fundus photograph showing a pigmented choroidal melanoma along the superotemporal arcade with a central nodule secondary to break through Bruch’s membrane, overlying lipofuscin and surrounding subretinal fluid (A). Autofluorescence demonstrates hyperautofluorescence overlying the lesion corresponding to lipofuscin (orange pigment) and surrounding shallow subretinal fluid (B). Ultra-widefield fundus photograph showing a circumpapillary pigmented choroidal melanoma (C). Autofluorescence demonstrates extensive subretinal fluid from 2 to 12 o’clock, which appears hyperautofluorescent (D). Ultra-widefield fundus photograph demonstrating a large choroidal melanoma overhanging and obscuring the optic nerve (E). Corresponding B-scan ultrasonography shows a typical mushroom-shape lesion with overlying retinal detachment (F). Ultra-widefield fundus photograph showing a temporal dome-shaped choroidal melanoma with B-scan ultrasonography demonstrating low/medium internal reflectivity of the lesion and associated exudative retinal detachment (G-H). |
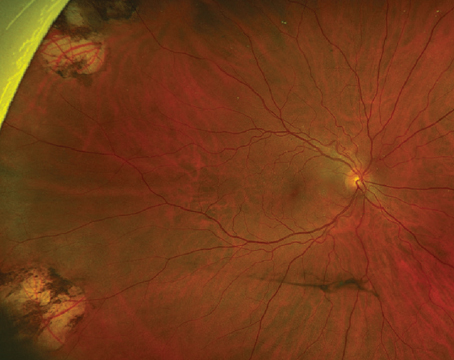
Choroidal melanoma is the most common primary intraocular malignancy in adults, occurring at an age-adjusted incidence of 4.3 per million in the United States.1 It usually presents in the sixth decade of life, most commonly as a pigmented lesion. In one large series, the mean largest basal dimension was 11.1 mm and average thickness was 5.5 mm.2 Clinical features supportive of a diagnosis of choroidal melanoma include the presence of subretinal fluid, presence of lipofuscin (orange pigment) associated with the lesion and rupture through Bruch’s membrane (Figure 1).
External and anterior segment examination is important to evaluate for the presence of ocular or oculodermal melanocytosis, a known risk factor for uveal melanoma, and for areas of possible extraocular extension. The presence of sentinel feeder vessels and/or asymmetric cataract could be associated with an underlying melanoma involving the ciliary body. On posterior segment examination, a multitude of factors in addition to location, size, shape and pigmentation of the lesion can help with the diagnosis. Choroidal melanomas are often dome-shaped and can also present in a mushroom-shape or with an overlying nodule if they’ve broken through Bruch’s membrane, which occurs in about 20 percent of cases.2
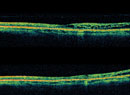
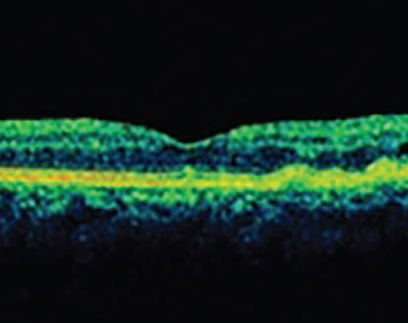
Given the systemic implications and the range of less concerning simulating lesions, multimodal imaging is extremely valuable in the correct identification of choroidal melanoma. Serial widefield color fundus photography can be particularly helpful when following indeterminate lesions for subtle evidence of growth, as well as monitoring treated choroidal melanomas for signs of local recurrence. Fundus autofluorescence (FAF) is another useful adjunct capable of confirming the presence of lipofuscin pigment as well as active or resolved areas of subretinal fluid, which can be subtle clinically but appear brightly hyperautofluorescent (Figure 1, B and D). Enhanced depth imaging optical coherence tomography (EDI-OCT) is helpful in identifying the contour of thinner tumors, the presence and extent of subretinal fluid, and changes of the overlying neurosensory retina. B-scan ultrasonography commonly shows a dome or mushroom-shaped lesion with medium to low internal reflectivity (Figure 1, F and H). B-scan ultrasonography is also critical in evaluating tumor thickness, spontaneous vascular pulsations, a sign of an intrinsic blood supply and the presence of extraocular extension.
Systemic screening is routinely performed for patients with choroidal melanoma to assess for distant metastasis, even though only 4 percent of patients are noted to have metastasis at the time of diagnosis.3 This workup is even more important in cases of diagnostic dilemmas in which a choroidal metastasis remains in the differential. When clinical examination and ancillary testing don’t clearly reveal the diagnosis, lesion biopsy, predominantly using fine-needle aspiration (FNAB) or a vitrector-assisted approach, can be performed. Biopsy is performed for cytopathology with or without immunohistochemistry, and more recently the DNA mutational profile can be ascertained to help establish the diagnosis using next-generation sequencing (NGS). On cytopathology, uveal melanoma demonstrates the presence of spindle and/or epithelioid cells often with the presence of cytoplasmic melanin.4 Immunohistochemical stains like SOX-10, HMB-45 and Melan-A confirm the melanocytic origin of the lesion, but don’t differentiate between a benign nevus and a malignant melanoma. Mitotic activity can be assessed using the Ki-67 stain, which can help distinguish these lesions and is associated with an increased risk of metastasis in uveal melanoma.5,6 Given the difficulty of cytopathologic differentiation, NGS may be helpful in obtaining an accurate diagnosis as melanocytic lesions are known to have a G-protein mutation in the majority of cases.7
The most common mutations include GNAQ, GNA11, PLCB4 and CYSLTR2, but less frequent initiator mutations are likely still to be discovered.3,7,8 Secondary mutations like BAP1, SF3B1 and EIF1AX are required for a lesion to have malignant potential, and their presence is indicative of a melanoma.3,9,10 The absence of either initial or secondary mutations doesn’t definitively rule out a melanocytic origin, and the absence of a secondary mutation in the presence of an initial mutation doesn’t definitively rule out a malignant lesion.3 NGS has not yet been studied prospectively, so its ultimate role in identification of tumor origin remains to be determined.
Choroidal Nevus
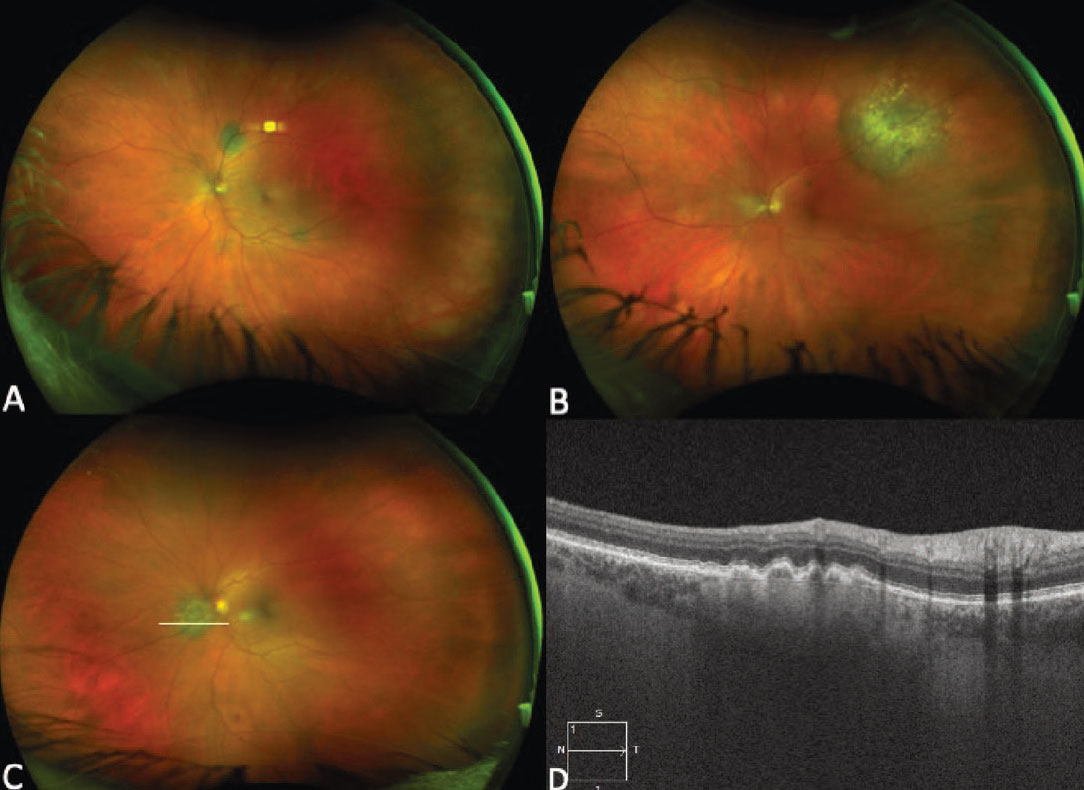 |
|
Figure 2. Ultra-widefield fundus photograph showing a small pigmented choroidal nevus superior to the optic nerve with overlying drusen, in addition to focal areas of pigment along the inferotemporal arcade (A). Ultra-widefield fundus photograph demonstrating a larger pigmented choroidal nevus along the superotemporal arcade, with prominent overlying drusen (B). Ultra-widefield fundus photograph and enhanced-depth imaging optical coherence tomography (EDI-OCT) demonstrating drusen overlying a peripapillary choroidal nevus (C and D). EDI-OCT also shows the limited thickness of the lesion, with posterior shadowing and loss of choroidal vascular details (D). |
Choroidal nevi are the most common intraocular tumors in adults. They are benign, acquired lesions that are typically discovered on routine dilated funduscopic examination.
The reported prevalence of choroidal nevi in the United States is 4.7 percent,11 but this may be an underestimation as many may go undetected due to peripheral location and lack of symptoms. They most commonly present as pigmented, flat or minimally elevated choroidal lesions, but some nevi have mixed pigmentation or are predominantly amelanotic. Although benign, choroidal nevi may cause symptoms if they are subfoveal, associated with subretinal fluid, or a choroidal neovascular membrane (CNVM) develops.12
While small, flat choroidal nevi are relatively easy to recognize, nevi that are larger, elevated and/or associated with high-risk features may be difficult to distinguish from small choroidal melanoma given the overlapping range of average dimensions, thickness and other clinical features.13,14 As such, multimodal imaging may assist in the diagnosis and management. On clinical examination, choroidal nevi are more likely to display signs of chronicity including overlying drusen, RPE atrophy and fibrous metaplasia, and can rarely be associated with a CNVM (Figure 2). The average nevus diameter was 1.25 mm in the Blue Mountains Eye Study and overlying drusen were present in 42 percent of lesions,15 but studies from tertiary referral centers have demonstrated larger average dimensions,16 likely because suspicious nevi are more likely to get referred to these practices. Multiple mnemonics have been developed to help clinicians risk-stratify melanocytic choroidal lesions, but no single feature can distinguish between a nevus and melanoma in isolation. The most commonly used mnemonic, To Find Small Ocular Melanoma (TFSOM), has been recently updated to To Find Small Ocular Melanoma Doing IMaging (TFSOM-DIM), incorporating multimodal imaging features. This stands for Thickness >2 mm (by ultrasound), Subretinal Fluid (on optical coherence tomography), Symptoms of vision loss (visual acuity <20/50 by Snellen acuity), Orange pigment (on fundus autofluorescence), Melanoma acoustic hollowness (on ultrasonography), and DIaMeter >5 mm (on fundus photography).17 This highlights the value of widefield color fundus photography, FAF, EDI-OCT and B-scan ultrasonography to identify meaningful risk factors.
Even when diagnosed accurately, a choroidal nevus must be monitored given the risk of malignant transformation to melanoma. The Cole Eye Institute’s Arun D. Singh, MD, and co-workers18 estimated the annual risk of malignant transformation of choroidal nevi at 1 in 8,845, while Wills Eye’s Carol Shields, MD, and co-authors19 reported a malignant transformation rate of 2, 9 and 13 percent of eyes at 1, 5 and 10 years in a cohort of 2,500 patients with many high-risk lesions. Lesions that have overlapping features of choroidal nevi and choroidal melanoma, sometimes referred to as “indeterminate melanocytic lesions” must be closely observed for signs of lesion growth.
In select cases, FNAB can be performed for cytopathologic diagnosis, as well as genetic mutational risk analysis of the lesion using gene expression profile testing or other molecular analysis. Cytopathology of choroidal nevi demonstrates polyhedral or spindle cells with small nuclei and lack of signs of cellular atypia.13
Congenital Hypertrophy of the Retinal Pigment Epithelium
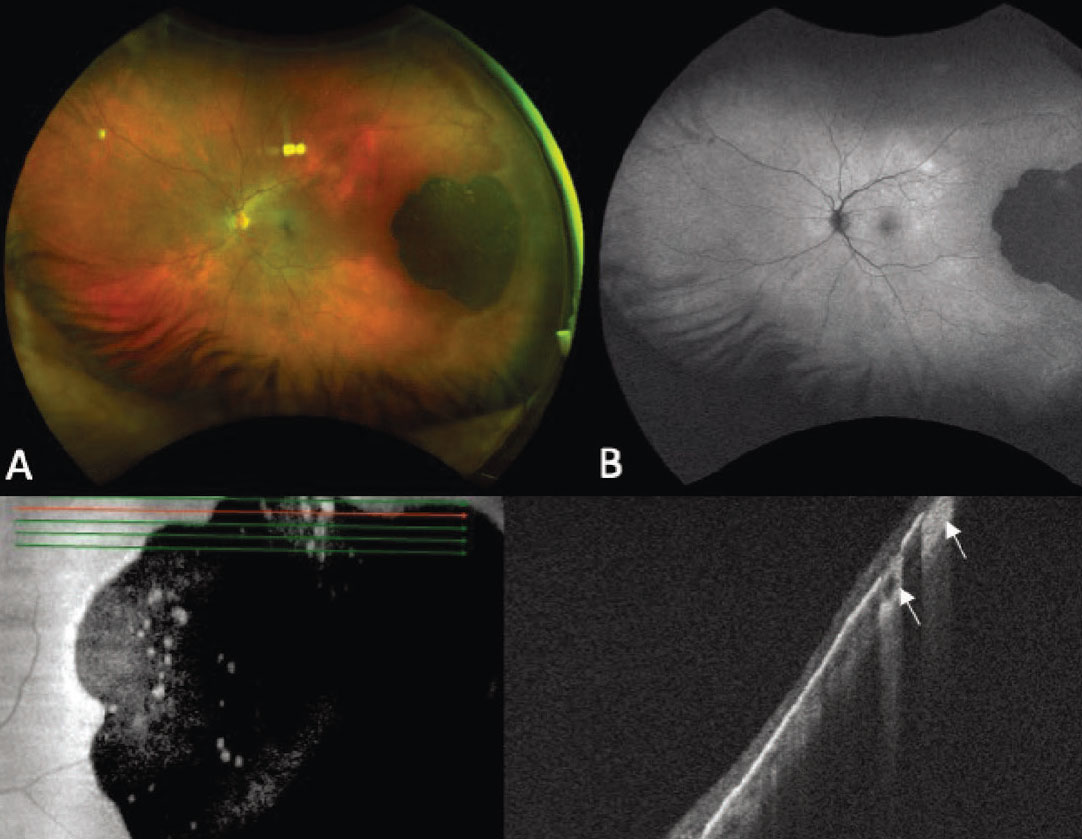 |
|
Figure 3. Ultra-widefield fundus photograph showing a flat, darkly pigmented congenital hypertrophy of the retinal pigment epithelium (CHRPE) with superior lacunae (A). Autofluorescence demonstrates hypoautofluorescence of the lesion with isoautofluorescence of the lacunae (B). Enhanced-depth imaging optical coherence tomography of the lesion shows hyperreflective RPE with thinning of the overlying neurosensory retina and RPE atrophy with increased signal transmission (white arrows), corresponding to the atrophic lacunae (C-D). |
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is a lesion of the RPE that most commonly presents as a flat, darkly pigmented lesion with or without lacunae (Figure 3, A).20 They tend to have geographic borders and are often located in the periphery. In a large series of 330 patients, 97 percent of patients presented without symptoms, and on B-scan ultrasonography the mean thickness of the lesions was 0.8 mm.20
The largest series of CHRPE demonstrated that these lesions can have benign growth with enlargement of basal dimensions over time.20 Rarely, CHRPE lesions can be associated with an RPE adenoma/adenocarcinoma, that appears as an elevated nodule in the context of CHRPE.20,21 Key clinical features distinguishing CHRPE from other pigmented choroidal lesions include lack of measurable thickness, dark pigmentation of the lesion and the presence of lacunae.20 FAF of CHRPE shows hypoauto-fluorescence of the lesion, with most lacunae demonstrating isoauto-fluorescence (Figure 3, B). OCT is helpful in confirming that the lesion is located in the RPE, demonstrated by thickening of the RPE, thinning/disorganization of the overlying neurosensory retina and an underlying choroid that is thin or normal in thickness.22,23 In areas of lacunae, OCT shows absence of RPE, allowing an increased transmission signal (Figure 3, D). Given the benign nature of CHRPE, these lesions are routinely observed.
Peripheral Exudative Hemorrhagic Chorioretinopathy
Peripheral exudative hemorrhagic chorioretinopathy (PEHCR) goes by a number of names, including extramacular disciform lesion and peripheral disciform pseudotumor. It’s characterized by hemorrhagic retinal degeneration, most commonly affecting the temporal, peripheral retina.24-26 While numerous descriptions of the disease process have been published in the literature, the term PEHCR was coined by Wills Eye’s William
Annesley Jr., MD, in 1980.25 In a series of “pseudomelanomas,” PEHCR was the second most frequent lesion referred for choroidal melanoma, behind choroidal nevi.27 PEHCR can manifest in a myriad of ways, but the presentation that most mimics choroidal melanomas is a focal pigmented elevated peripheral lesion. Choroidal melanoma was the referring diagnosis in 99 and 41 percent of cases ultimately diagnosed as PEHCR at an ocular oncology tertiary referral center and a series of non-ocular oncology practices, respectively, reinforcing the diagnostic uncertainty.24,26
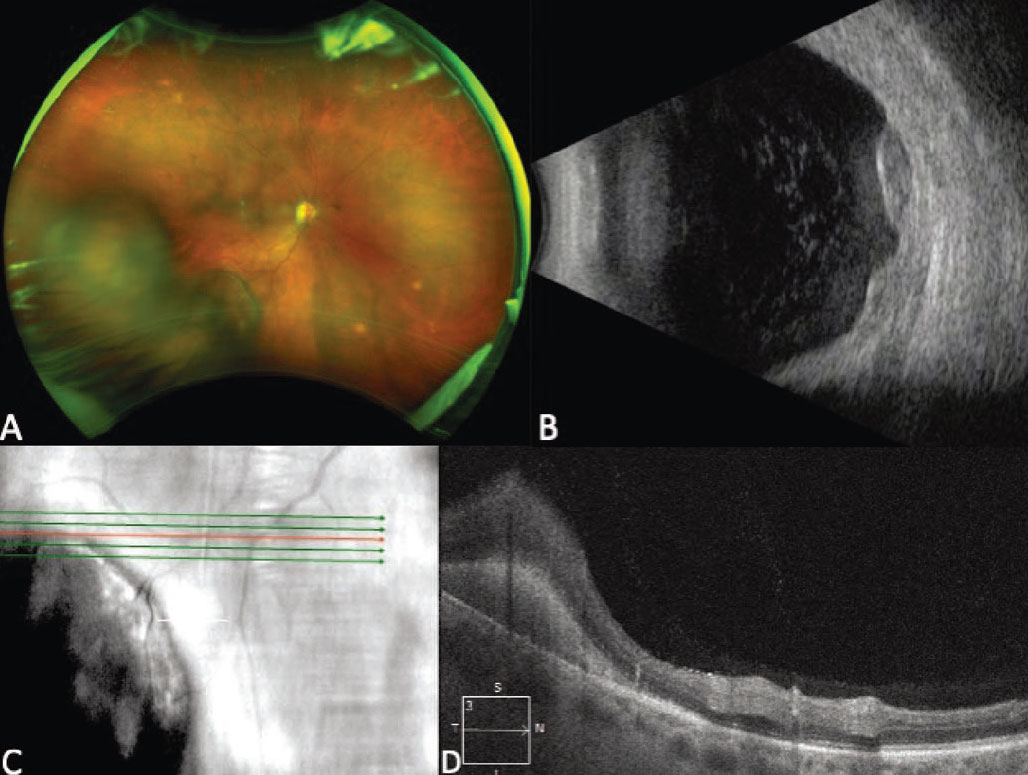 |
|
Figure 4. Ultra-widefield fundus photograph showing a multilobular pigmented lesion in the inferotemporal periphery consistent with peripheral exudative hemorrhagic chorioretinopathy (PEHCR), in a patient on apixaban and clopidogrel (A). B-scan ultrasonography of the lesion demonstrates a multilobular lesion with an irregular surface and heterogeneous internal reflectivity (B). Enhanced-depth imaging optical coherence tomography at the posterior margin of the lesion shows hyperreflectivity in the subretinal space consistent with subretinal hemorrhage (C-D). |
PEHCR most commonly presents in the seventh and eighth decades of life, occurs in Caucasians in the vast majority of cases, and has a female predominance.24,26,28 Clinically, PEHCR presents as a focal hemorrhagic lesion, or with more diffuse exudation and/or hemorrhage (Figure 4, A). Vitreous, subretinal and sub-RPE hemorrhage can occur with PEHCR, and active simultaneous, bilateral disease occurs in 21 to 43 percent of cases.24,26,28,29 This may be an underestimation of the actual prevalence of bilateral disease, as patients may have chorioretinal pigmentary changes in the “uninvolved” eye, indicative of previous active disease. Bilaterality is an important distinguishing feature of PEHCR, as bilateral choroidal melanoma is incredibly rare.28 When present, exudation near the lesion is another distinguishing feature of PEHCR, as choroidal melanoma isn’t typically associated with exudation prior to treatment. A medical history, review of systems and medication evaluation can be helpful in obtaining a diagnosis, as 9 to 44 percent of people with PEHCR are on anticoagulation.24,26 Close observation may be another helpful diagnostic tool as lesion regression or stability occurs without intervention in nearly 90 percent of cases.24
On indocyanine green angiography, peripheral polypoidal dilations are common.26,28,29 On fluorescein angiography, hypofluorescence from blockage is often seen secondary to hemorrhage. Late irregular hyperfluorescence is also common while obvious CNVMs are less frequently visualized. 24,26,29 Studies of OCT features in PEHCR demonstrate the presence of pigment epithelial detachments in many cases and have also shown increased choroidal thickness temporal to the fovea compared to control eyes.28,30 Peripheral OCT of the posterior border of the lesion can also be helpful if visualization is sufficient to localize the lesion to the subretinal or sub-RPE space (Figure 4D).28 On B-scan ultrasonography, the average thickness of PEHCR lesions is around 3 mm, and the lesions may demonstrate a multilobulated, dome, plateau or irregular shape (Figure 4B).24,29 PEHCR lesions don’t exhibit spontaneous vascular pulsations and often have heterogenous internal acoustic characteristics.26 In the setting of significant hemorrhage preventing visualization of the lesion, repeat ultrasonography is critical to rule out increased lesion thickness and a mushroom configuration suggestive of a melanoma.
Choroidal Melanocytosis
Choroidal melanocytosis is a congenital hyperpigmentation of the choroid that most commonly presents as a unilateral sectoral or diffuse hyperpigmentation.31 Choroidal melanocytosis can occur in isolation or in association with ocular/oculodermal melanocytosis.31-33
Demographics and clinical examination are helpful in distinguishing choroidal melanocytosis from other pigmented lesions. It’s thought to be a congenital lesion and is often diagnosed at an earlier age than most other pigmented choroidal lesions.31 External and anterior segment examination is critical to detect periocular, scleral and/or iris hyperpigmentation. On fundoscopic examination, there’s no appreciable choroidal thickening of the lesion. Additionally, the absence of drusen and chronic RPE changes helps to distinguish the lesion from broad-based choroidal nevi, and the absence of lipofuscin and subretinal fluid can help distinguish the lesion from a diffuse choroidal melanoma.31 The risk of uveal melanoma development in Caucasian patients with ocular or oculodermal melanocytosis is 1/400.34,35 The risk of developing uveal melanoma in patients with isolated choroidal melanocytosis is not known, but appears to be increased.31,32 This increased risk of uveal melanoma warrants lifetime monitoring to detect melanoma early (Figure 5).
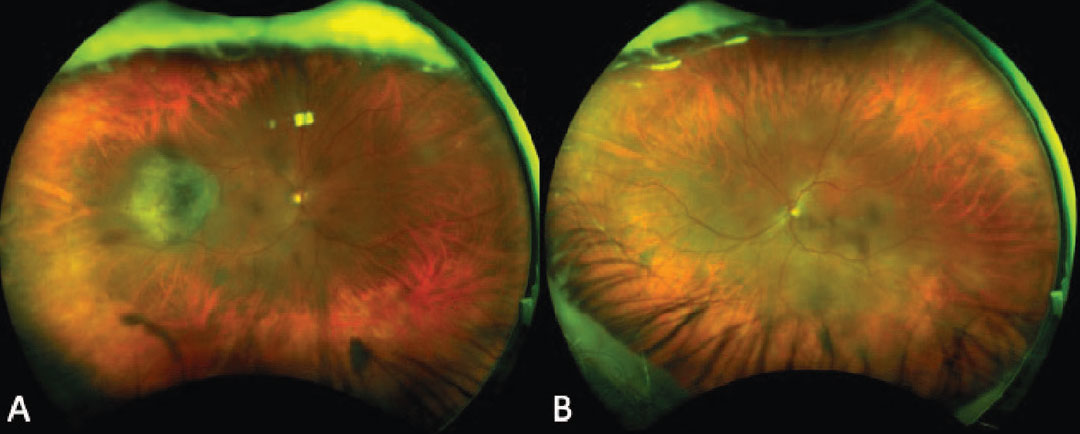 |
|
Figure 5. Ultra-widefield fundus photograph of a patient with bilateral isolated choroidal melanocytosis who developed an associated elevated, pigmented choroidal melanoma in the temporal midperiphery of the right eye that subsequently underwent plaque brachytherapy (A-B). |
A combination of serial clinical examination, widefield color fundus photography, FAF, EDI-OCT, B-scan ultrasonography and ultrasound biomicroscopy can help detect the development of uveal melanoma at its earliest stage. FAF of choroidal melanocytosis shows isoautofluorescence of the lesion and is useful to rule out the presence of lipofuscin that could suggest an accompanying choroidal melanoma.36 EDI-OCT of choroidal melanocytosis shows slight choroidal thickening compared to the adjacent non-affected choroid while B-scan ultrasonography demonstrates no appreciable thickening.37 Significant thickening or subretinal fluid seen on EDI-OCT or ultrasonography should raise suspicion for choroidal melanoma or choroidal nevus.
Pigmented Choroidal Metastasis
While choroidal metastasis from carcinoma usually presents as an amelanotic lesion, metastasis from primary cutaneous or uveal melanoma may appear as a pigmented lesion.38-40 Additionally, rare cases of pigmented choroidal metastasis from a neuroendocrine tumor and lung adenocarcinoma have been published.41,42 In a large series of 1,111 patients with uveal metastasis by Dr. Shields’ group, cutaneous melanoma was the fifth most common known primary tumor at 2 percent of cases.38 Uveal metastasis from cutaneous melanoma occurred at a mean age of 59 years, all of the patients were Caucasian, and 96 percent had a known diagnosis of cutaneous melanoma prior to the diagnosis of uveal metastasis.38
Cases suspicious for choroidal metastasis warrant a detailed anterior and posterior segment exam to identify other metastatic lesions in either eye. Metastatic cutaneous melanoma also has a propensity for vitreous involvement, which has been previously reported to rarely occur simultaneously with a pigmented choroidal lesion.43 Pigmented choroidal metastatic lesions can also rarely occur from metastatic choroidal melanoma in the same or fellow eye. Tehran’s Babak Masoomian, MD, and his fellow investigators40 studied 13 patients with choroidal or ciliary body melanoma with metastasis to the contralateral ocular and periocular structures, out of 13,000 total uveal melanoma cases. In their study, four patients demonstrated choroidal metastasis in the fellow eye, with 3/4 demonstrating multifocal choroidal lesions. The rate of contralateral metastasis of choroidal/ciliary body melanoma was 13/13,000 (0.1 percent) and 11 of these patients (85 percent) demonstrated evidence of systemic metastasis.40 Metastatic choroidal melanoma has also been reported to metastasize to the ipsilateral and/or contralateral orbit or eyelid, and can present with new-onset diplopia, in addition to choroidal involvement.40
On clinical examination, choroidal metastases from cutaneous or choroidal melanoma typically present as dome- or plaque-shaped pigmented choroidal lesions. These lesions may be multifocal and can be bilateral, but can also present as unifocal.38 The presence of multifocal pigmented lesions should raise concern of metastasis (Figure 6, A and B). Extensive subretinal fluid and the presence of lipofuscin can accompany these tumors, thus making these lesions especially difficult to distinguish from primary choroidal melanoma.38
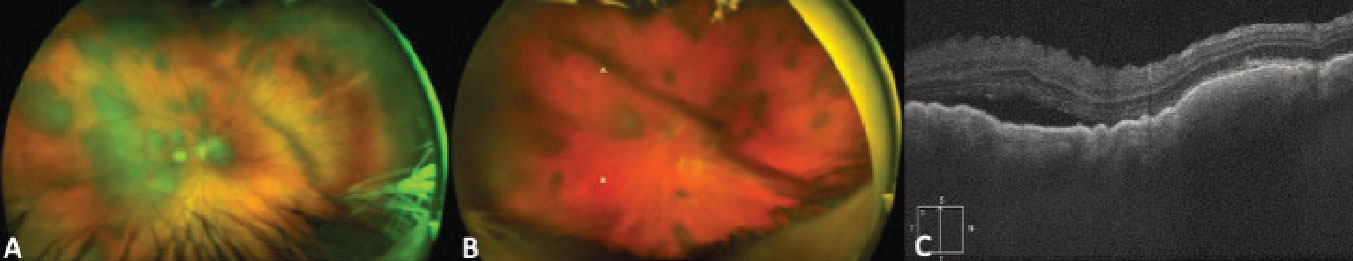 |
|
Figure 6. Ultra-widefield fundus photograph of the right (A) and left (B) eyes demonstrating bilateral multifocal choroidal metastasis in a patient with metastatic cutaneous melanoma. Enhanced-depth imaging optical coherence tomography vertical raster scan over the macula of the right eye showing an undulating surface of the lesion with choroidal thickening and associated subretinal fluid (C). |
In addition to widefield color fundus photographs demonstrating pigmented unifocal or multifocal lesions, FAF can be used to confirm lipofuscin when present. EDI-OCT can be very helpful in identifying metastatic lesions, as they often present with an undulating appearance and more subretinal fluid than a choroidal melanoma of similar size. (Figure 6, C). Some primary choroidal melanomas can also present with an undulating appearance, so this configuration isn’t pathognomonic. B-scan ultrasonography typically demonstrates an acoustically dense lesion with high internal reflectivity compared to primary choroidal melanoma, which more commonly has medium to low internal reflectivity. Given the clinical and imaging overlap between metastatic melanoma and primary choroidal melanoma, a known history of previous choroidal melanoma or skin melanoma is especially important to elicit.
Melanocytoma
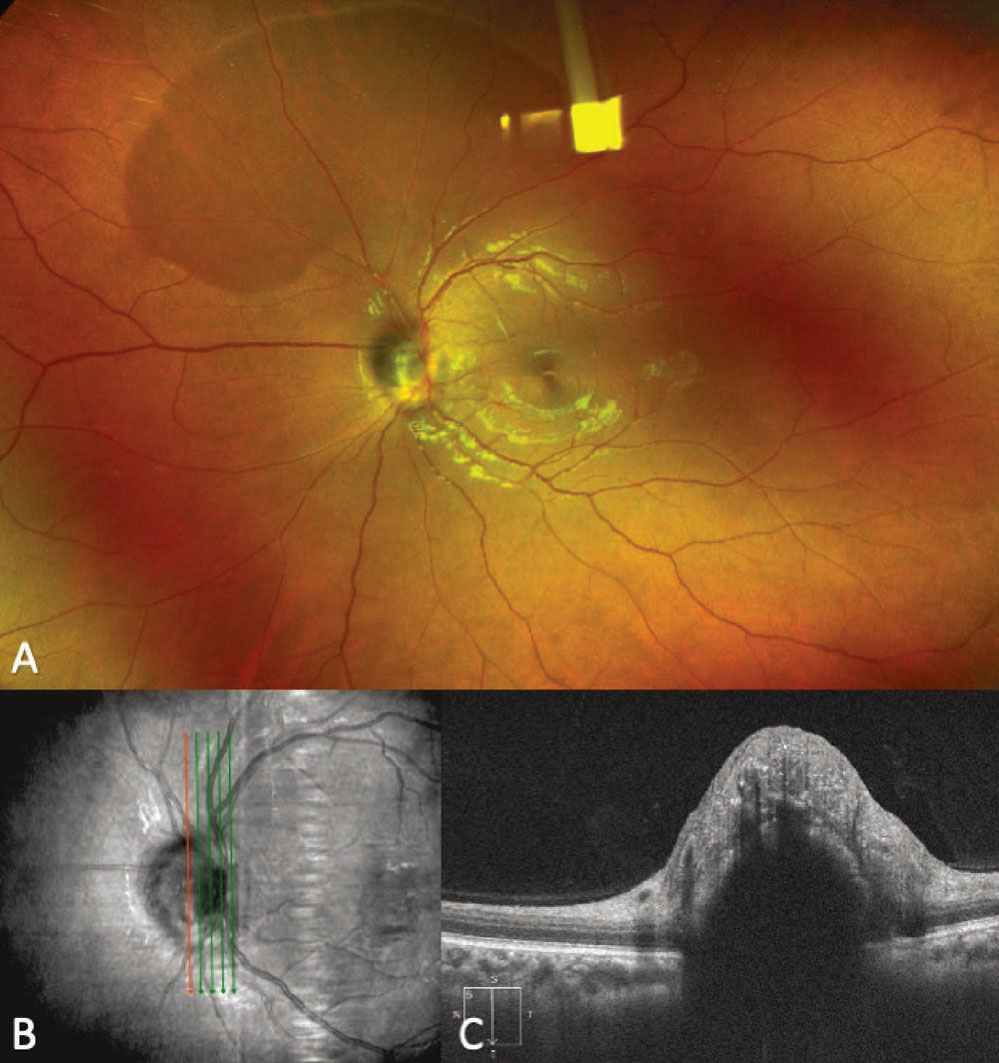 |
|
Figure 7. Fundus photograph showing a melanocytoma of the optic disc (A). An enhanced-depth imaging optical coherence tomography vertical raster scan overlying the optic disc demonstrates an elevated lesion with disorganization of the overlying retina and posterior shadowing (B-C). |
Melanocytoma is a darkly pigmented, benign variant of a melanocytic nevus with a low risk of malignant transformation.44 These lesions may be located anywhere in the posterior segment but are most commonly located at the optic nerve. Prior to the landmark paper by Lorenz Zimmerman45 in 1962, melanocytomas were commonly thought to be malignant lesions requiring enucleation. Following that histopathological investigation demonstrating the benign nature of these lesions, conservative management was adopted.45 Demographically, melanocytomas are more likely to occur in African-American patients than choroidal nevi or choroidal melanomas. Wills Eye’s Jerry Shields, MD, and co-workers44 reported 29 percent of melanocytoma cases occurring in African Americans, as compared to less than 1 percent of patients with uveal melanoma.2
Key clinical findings of melanocytomas include a unilateral, darkly pigmented lesion, most commonly overlying the optic disc, with or without extension to the adjacent retina or choroid (Figure 7, A).44 While often an incidental finding, melanocytoma, especially when located at the optic disc, can cause blurred vision; visual field defects; afferent pupillary defects; disc edema; and intraretinal and subretinal edema.44,46-48 Less commonly, melanocytoma of the optic disc can present with tumor necrosis or central retinal vein occlusions.44,47 Clinically, melanocytomas tend to be more darkly pigmented than choroidal melanomas. They also may overlie the optic disc, while melanomas can abut or surround the disc and don’t typically primarily overlie the disc unless they are larger in size than most melanocytomas.
While clinical examination is often sufficient for making the diagnosis of melanocytoma, multimodal imaging can help in cases of diagnostic ambiguity. FAF demonstrates hypoautofluorescence of the lesion.49 OCT of melanocytomas most typically demonstrates an elevated lesion with a hyperreflective anterior surface, posterior shadowing and a disorganized overlying retina, although other OCT patterns have been described (Figure 7, B).50,51 B-scan ultrasonography and color fundus photography can be performed to document the lesion size and clinical appearance for future comparison, but often don’t help differentiate melanocytomas from other pigmented posterior segment lesions.
Bilateral Diffuse Uveal Melanocytic Proliferation
Bilateral diffuse uveal melanocytic proliferation (BDUMP) is a rare ocular paraneoplastic syndrome most commonly associated with urogenital cancer in women and lung carcinomas in men, although a variety of associated malignancies have been reported, typically occurring in the sixth to eighth decades of life.52 BDUMP was first described by Robert Machemer, MD, in 1966,53 and J. Donald M. Gass, MD, described its five cardinal features, including focal areas of red pigmentation at the level of the RPE, multifocal hyperfluorescence of these focal red patches, multiple focal pigmented and nonpigmented uveal lesions with slight elevation, exudative retinal detachment and rapidly progressing cataracts.54 Patients typically present with bilateral progressive vision loss associated with exudative retinal detachment, outer retina disruption and cataracts. This is the first indication of systemic malignancy in greater than 40 percent of patients.52
The multiple, minimally elevated, pigmented lesions of BDUMP are unlikely to be confused with choroidal melanoma given their multifocality, size and minimal elevation, but could simulate multifocal choroidal nevi, choroidal metastases from skin or uveal melanoma, or the multifocal pigmented lesions seen in Gardner syndrome.
FAF highlights the typical pattern of both hypo- and hyperautofluorescent areas, corresponding to RPE atrophy and hyperplasia. EDI-OCT and B-scan ultrasonography are also helpful ancillary tests in patients with suspected BDUMP as they can document and measure the uveal thickening. Additionally, UBM can be performed to identify iris pigment epithelium and/or ciliary body cysts, which have been described in some cases of BDUMP. A detailed history should address any previously known systemic malignancy and include a thorough review of systems to identify signs of an undetected malignancy. Systemic imaging should be performed in the absence of known systemic cancer or if the patient is presumed to be in remission but hasn’t had a recent evaluation.
Retinal Pigment Epithelium Adenoma/Adenocarcinoma
Retinal pigment epithelium adenoma/adenocarcinoma is a very rare tumor of the RPE that classically presents as a darkly pigmented, abruptly elevated, unilateral, nodular lesion.21 Given the clinical similarities, these lesions are commonly misdiagnosed as choroidal melanoma. Because RPE adenomas/adenocarcinomas are retinal lesions, they may have feeding arteries (extremely rare in choroidal melanoma) and draining veins (not previously reported in melanoma). Vitreous seeding may also be seen with RPE adenomas/adenocarcinomas, but is rare for choroidal melanomas unless they’ve broken through Bruch’s membrane and invaded the retina. Another differentiating factor is surrounding or distant lipid exudation, a feature not appreciated in patients with untreated choroidal melanoma. Lastly, RPE adenomas/adenocarcinomas are associated with an underlying CHRPE in 22 percent of cases. This is an extremely rare finding in choroidal melanomas and is presumed to be incidental.21,55
Widefield color fundus photography can help document the darkly pigmented nodular lesion. FA shows early hypofluorescence with late iso- or hyperfluorescence, often with leakage or staining. EDI-OCT evaluation of thinner lesions, or at the margin of thicker lesions, documents localization to the RPE. It often demonstrates high reflectivity, retinal invasion and a “derby hat” appearance with a vertical elevation at the edge of the lesion.21 B-scan ultrasonography shows dome-shaped or abruptly elevated “derby hat” lesions, typically with medium or high internal reflectivity.21 Distinguishing between RPE adenoma/adenocarcinoma and choroidal melanoma is particularly important given the prognostic implication, as RPE adenoma/adenocarcinoma has only been reported to metastasize in a single case in the literature.56
If FNAB is required for diagnostic confirmation, cytopathology demonstrates pleomorphic pigmented cells in a solid or vacuolar pattern with large melanosomes, compared to spindle cells with smaller melanosomes seen more commonly in uveal melanoma. Immunohistochemistry results are typically positive for CAM 5.2, AE1/AE3 and vimentin in most cases, but can also overlap with uveal melanoma results and be positive for HMB-45, Melan-A and S-100 in some cases.21 Cytopathology must be viewed in light of the clinical examination as results of RPE adenoma/adenocarcinoma FNAB have been misdiagnosed as other lesions prior to enucleation revealing the correct diagnosis.21
Suprachoroidal Hemorrhage
Suprachoroidal hemorrhage is a rare entity thought to occur secondary to rupture of a posterior ciliary artery associated with hypotony, systemic vascular risk factors, anticoagulation and at-risk choroidal vasculature. While most commonly occurring in the intraoperative or immediate postoperative period, spontaneous suprachoroidal hemorrhage secondary to Valsalva can rarely occur.57 There tends not to be diagnostic confusion in the setting of diffuse hemorrhage, but a localized hemorrhage can appear as a focal pigmented lesion and be mistaken for a choroidal melanoma.57,58 One subtle clinical finding often seen with suprachoroidal hemorrhage is overlying radial choroidal folds. Suprachoroidal hemorrhage also tends to have more ill-defined margins than choroidal melanoma or nevus (Figure 8, A).
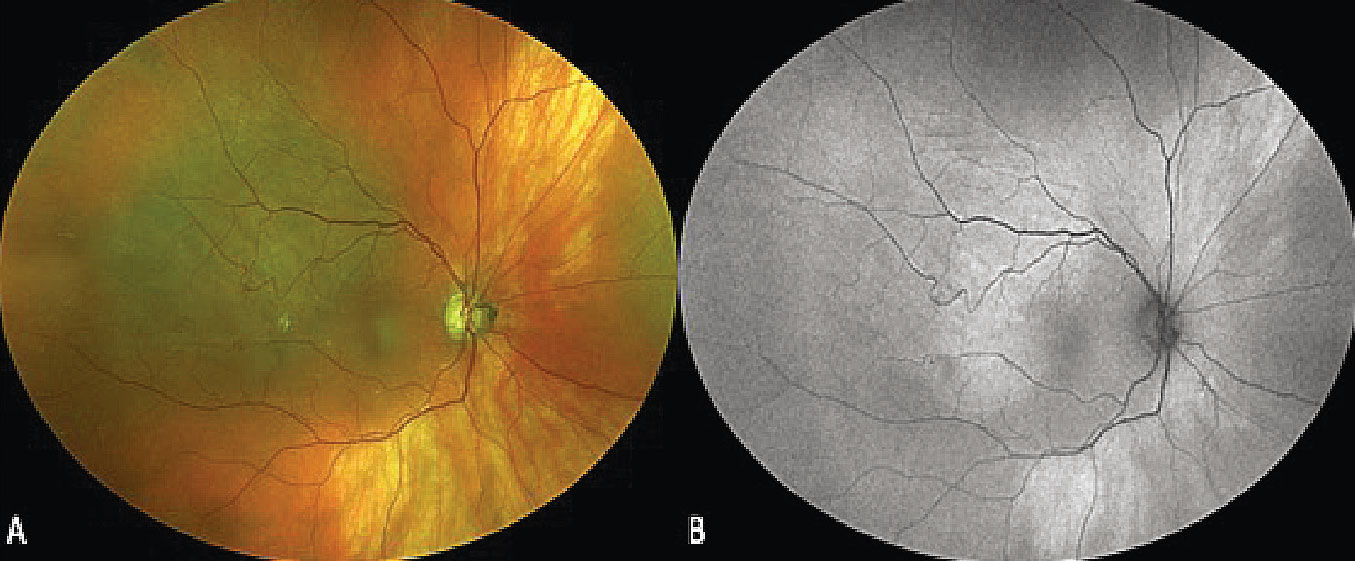 |
|
Figure 8. Ultra-widefield fundus photograph demonstrating a superotemporal pigmented choroidal lesion with ill-defined borders and overlying choroidal folds (A). Fundus autofluorescence shows isoautofluorescence of the lesion with minimal hyperautofluorescence along the posterior margin. The overlying choroidal folds are visible as faint, hypoautofluorescent linear streaks (B). |
History is important in distinguishing the lesions, as most suprachoroidal hemorrhages occur in the intraoperative and postoperative course and have been associated with filtering surgery, cataract surgery, keratoplasty and pars plana vitrectomy at differing rates.59,60 In addition to recent surgery, patients may present with a dull pain that is uncommon with choroidal melanoma unless associated with necrotic tumors and scleritis. Typically, localized suprachoroidal hemorrhages will show some degree of resolution within weeks to months, so close observation may also reveal the diagnosis.
Multimodal imaging is helpful in diagnosing a localized suprachoroidal hemorrhage. FAF typically shows isoautofluorescence in the area of the lesion as the overlying RPE is often intact, but the choroidal folds may be well highlighted as linear, mildly hyper- or hypoautofluorescent streaks (Figure 8, B). Both FA and ICGA demonstrate a normal retinal and choroidal circulation overlying the area of hemorrhage and can show areas of hypofluorescence from choroidal folds.61 EDI-OCT of thinner suprachoroidal hemorrhages can be critical to the diagnosis by documenting the hyporeflective hemorrhage being located between the sclera and the choroid. B-scan cuts oriented perpendicular to choroidal folds, if present, can also be documented on OCT.57 B-scan ultrasonography may demonstrate heterogenous internal reflectivity, and lesion thickness varies based on the amount of hemorrhage.60
In conclusion, a multitude of pigmented choroidal lesions and simulating conditions exist, and overlapping or atypical clinical features can result in diagnostic confusion. Clinical examination is paramount, but patient history and multimodal imaging may be critical in narrowing the differential diagnosis to distinguish and properly manage these lesions.
Correspondence:
Basil K. Williams Jr., MD
Ocular Oncology Service, University of Cincinnati College of Medicine
231 Albert Sabin Way, Suite 5414
Cincinnati, OH 45267-0527.
Tel: (513) 475-7300
Fax (513) 475-7311
Email: willi5b2@ucmail.uc.edu
1. Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology 2003;110:5:956-61.
2. Shields CL, Kaliki S, Furuta M, et al. Clinical spectrum and prognosis of uveal melanoma based on age at presentation in 8,033 cases. Retina 2012;32:7:1363-72.
3. Smit KN, Jager MJ, de Klein A, Kili E. Uveal melanoma: Towards a molecular understanding. Prog Retin Eye Res 2020;75:100800.
4. Medina CA, Biscotti CV, Singh N, Singh AD. Diagnostic cytologic features of uveal melanoma. Ophthalmology 2015;122:8:1580-4.
5. Onken MD, Worley LA, Harbour JW. Association between gene expression profile, proliferation and metastasis in uveal melanoma. Curr Eye Res 2010;35:9:857-63.
6. Staby KM, Gravdal K, Mørk SJ, et al. Prognostic impact of chromosomal aberrations and GNAQ, GNA11 and BAP1 mutations in uveal melanoma. Acta Ophthalmol 2018;96:1:31-38.
7. Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med 2010;363:23:2191-9.
8. Moore AR, Ceraudo E, Sher JJ, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet 2016;48:6:675-80.
9. Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010;330:6009:1410-3.
10. Yavuzyigitoglu S, Koopmans AE, Verdijk RM, et al. Uveal melanomas with SF3B1 mutations: A distinct subclass associated with late-onset metastases. Ophthalmology 2016;123:5:1118-28.
11. Qiu M, Shields CL. Choroidal nevus in the united states adult population: Racial disparities and associated factors in the National Health and Nutrition Examination Survey. Ophthalmology 2015;122:10:2071-83.
12. Chien JL, Sioufi K, Surakiatchanukul T, et al. Choroidal nevus: A review of prevalence, features, genetics, risks, and outcomes. Curr Opin Ophthalmol 2017;28:3:228-237.
13. Gass JD. Problems in the differential diagnosis of choroidal nevi and malignant melanomas. The XXXIII Edward Jackson Memorial Lecture. Am J Ophthalmol 1977;83:3:299-323.
14. Augsburger JJ, Corrêa ZM, Trichopoulos N, Shaikh A. Size overlap between benign melanocytic choroidal nevi and choroidal malignant melanomas. Invest Ophthalmol Vis Sci 2008;49:7:2823-8.
15. Sumich P, Mitchell P, Wang JJ. Choroidal nevi in a white population: The Blue Mountains Eye Study. Arch Ophthalmol 1998;116:5:645-50.
16. Shields CL, Furuta M, Mashayekhi A, et al. Clinical spectrum of choroidal nevi based on age at presentation in 3422 consecutive eyes. Ophthalmology 2008;115:3:546-552. e2.
17. Shields CL, Dalvin LA, Ancona-Lezama D, et al. Choroidal nevus imaging features in 3,806 cases and risk factors for transformation into melanoma in 2,355 cases: The 2020 Taylor R. Smith and Victor T. Curtin Lecture. Retina 2019;39:10:1840-1851.
18. Singh AD, Kalyani P, Topham A. Estimating the risk of malignant transformation of a choroidal nevus. Ophthalmology 2005;112:10:1784-9.
19. Shields CL, Furuta M, Berman EL, et al. Choroidal nevus transformation into melanoma: Analysis of 2,514 consecutive cases. Arch Ophthalmol 2009;127:8:981-7.
20. Shields CL, Mashayekhi A, Ho T, et al. Solitary congenital hypertrophy of the retinal pigment epithelium: Clinical features and frequency of enlargement in 330 patients. Ophthalmology 2003;110:10:1968-76.
21. Williams BK Jr, Di Nicola M, Acaba-Berrocal LA, et al. Adenoma and adenocarcinoma of the retinal pigment epithelium: A review of 51 consecutive patients. Ophthalmol Retina 2020;4:8:829-839.
22. Francis JH, Sobol EK, Greenberg M, et al. Optical coherence tomography characteristics of the choroid underlying congenital hypertrophy of the retinal pigment epithelium. Ocul Oncol Pathol 2020;6:4:238-243.
23. Fung AT, Pellegrini M, Shields CL. Congenital hypertrophy of the retinal pigment epithelium: enhanced-depth imaging optical coherence tomography in 18 cases. Ophthalmology 2014;121:1:251-256.
24. Shields CL, Salazar PF, Mashayekhi A, Shields JA. Peripheral exudative hemorrhagic chorioretinopathy simulating choroidal melanoma in 173 eyes. Ophthalmology 2009;116:3:529-35.
25. Annesley WH, Jr. Peripheral exudative hemorrhagic chorioretinopathy. Trans Am Ophthalmol Soc 1980;78:321.
26. Vandefonteyne S, Caujolle JP, Rosier L, et al. Diagnosis and treatment of peripheral exudative haemorrhagic chorioretinopathy. Br J Ophthalmol 2020;104:6:874-878.
27. Shields CL, Manalac J, Das C, Ferguson K, Shields JA. Choroidal melanoma: Clinical features, classification, and top 10 pseudomelanomas. Curr Opin Ophthalmol 2014;25:3:177-85.
28. Zicarelli F, Preziosa C, Staurenghi G, Pellegrini M. Peripheral exudative haemorrhagic chorioretinopathy: A widefield imaging study. Br J Ophthalmol 2021;105:10:1410.
29. Mantel I, Uffer S, Zografos L. Peripheral exudative hemorrhagic chorioretinopathy: a clinical, angiographic, and histologic study. Am J Ophthalmol 2009;148:6:932-8.e1.
30. Shroff D, Sharma M, Chhablani J, et al. Peripheral exudative hemorrhagic chorioretinopathy-A new addition to the spectrum of pachychoroid disease? Retina 2021;41:7:1518-1525.
31. Augsburger JJ, Brooks CC, Correa ZM. Isolated choroidal melanocytosis: Clinical update on 37 cases. Graefes Arch Clin Exp Ophthalmol 2020;258:12:2819-2829.
32. Augsburger JJ, Trichopoulos N, Corrêa ZM, Hershberger V. Isolated choroidal melanocytosis: A distinct clinical entity? Graefes Arch Clin Exp Ophthalmol 2006;244:11:1522-7.
33. Shields CL, Kaliki S, Livesey M, et al. Association of ocular and oculodermal melanocytosis with the rate of uveal melanoma metastasis: Analysis of 7872 consecutive eyes. JAMA Ophthalmol 2013;131:8:993-1003.
34. Singh AD, De Potter P, Fijal BA, et al. Lifetime prevalence of uveal melanoma in white patients with oculo(dermal) melanocytosis. Ophthalmology 1998;105:1:195-8.
35. Teekhasaenee C, Ritch R, Rutnin U, Leelawongs N. Ocular findings in oculodermal melanocytosis. Archives of ophthalmology 1990;108:8:1114-20.
36. Hrynchak P, Hugh J, Labreche T. Bilateral isolated choroidal melanocytosis with isoautofluorescence. Can J Ophthalmol 2018;53:3:e97-e99.
37. Pellegrini M, Shields CL, Arepalli S, Shields JA. Choroidal melanocytosis evaluation with enhanced depth imaging optical coherence tomography. Ophthalmology 2014;121:1:257-261.
38. Shields CL, Welch RJ, Malik K, et al. Uveal metastasis: Clinical features and survival outcome of 2214 tumors in 1111 patients based on primary tumor origin. Middle East Afr J Ophthalmol 2018;25:2:81-90.
39. Font RL, Naumann G, Zimmerman LE. Primary malignant melanoma of the skin metastatic to the eye and orbit. Report of ten cases and review of the literature. Am J Ophthalmol 1967;63:4:738-54.
40. Masoomian B, Mashayekhi A, Shields JA, Shields CL. Uveal melanoma metastasis to the contralateral eye structures: A retrospective comparative analysis of 13 consecutive patients. Ophthalmol Retina 2021;5:10:1036.
41. Eagle RC, Jr., Ehya H, Shields JA, Shields CL. Choroidal metastasis as the initial manifestation of a pigmented neuroendocrine tumor. Arch Ophthalmol 2000;118:6:841.
42. Frenkel S, Pe’er J. Choroidal metastasis of adenocarcinoma of the lung presenting as pigmented choroidal tumor. Case Rep Ophthalmol 2012;3:3:311-6.
43. Francis JH, Berry D, Abramson DH, et al. Intravitreous cutaneous metastatic melanoma in the era of checkpoint inhibition: Unmasking and masquerading. Ophthalmology 2020;127:2:240-248.
44. Shields JA, Demirci H, Mashayekhi A, Shields CL. Melanocytoma of optic disc in 115 cases: The 2004 Samuel Johnson Memorial Lecture, part 1. Ophthalmology 2004;111:9:1739-46.
45. Zimmerman LE, Garron LK. Melanocytoma of the optic disc. International Ophthalmology Clinics 1962;2:2:431.
46. Osher RH, Shields JA, Layman PR. Pupillary and visual field evaluation in patients with melanocytoma of the optic disc. Arch Ophthalmol 1979;97:6:1096-9.
47. Shields JA, Demirci H, Mashayekhi A, Eagle RC, Jr., Shields CL. Melanocytoma of the optic disk: a review. Surv Ophthalmol 2006;51:2:93-104.
48. Joffe L, Shields JA, Osher RH, Gass JD. Clinical and follow-up studies of melanocytomas of the optic disc. Ophthalmology 1979;86:6:1067-83.
49. Salvanos P, Utheim TP, Moe MC. Autofluorescence imaging in the differential diagnosis of optic disc melanocytoma. Acta Ophthalmol 2015;93:5:476-80.
50. Shields CL, Perez B, Benavides R, et al. Optical coherence tomography of optic disk melanocytoma in 15 cases. Retina 2008;28:3:441-6.
51. Apinyawasisuk S, McCannel T, Arnold AC. Clinical and spectral-domain optical coherence tomography appearance of optic disc melanocytoma: A new classification and differentiation from pigmented choroidal lesions. Ocul Oncol Pathol 2017;3:2:142-148.
52. Klemp K, Kiilgaard JF, Heegaard S. Bilateral diffuse uveal melanocytic proliferation: Case report and literature review. Acta Ophthalmol 2017;95:5:439-445.
53. Machemer R. [On the pathogenesis of the flat malignant melanoma]. Klin Monbl Augenheilkd 1966;148:5:641-52.
54. Gass JD, Gieser RG, Wilkinson CP. Bilateral diffuse uveal melanocytic proliferation in patients with occult carcinoma. Arch Ophthalmol 1990;108:4:527-33.
55. Williams BK Jr, Di Nicola M, Lucio-Alvarez JA, et al. Choroidal melanoma simulating adenoma of the retinal pigment epithelium arising at the site of congenital hypertrophy of the retinal pigment epithelium. Ocul Oncol Pathol 2020;6:1:39-43.
56. Heindl LM, Naumann GO, Kruse FE, Holbach LM. Aggressive metastasising adenocarcinoma of the retinal pigment epithelium with trisomy 21. The British journal of ophthalmology 2008;92:3:389-91.
57. Marous CL, Sioufi K, Shields CL. Coughing-induced suprachoroidal hemorrhage simulating melanoma in two cases. Retin Cases Brief Rep 2018;12:4:336-341.
58. Honig SE, Srinivasan A, Shields CL. Suprachoroidal hemorrhage simulating melanoma in idiopathic thrombocytopenic purpura. Ocul Oncol Pathol 2019;5:3:162-166.
59. Welch JC, Spaeth GL, Benson WE. Massive suprachoroidal hemorrhage. Follow-up and outcome of 30 cases. Ophthalmology 1988;95:9:1202-6.
60. Mantopoulos D, Hariprasad SM, Fine HF. Suprachoroidal hemorrhage: Risk factors and diagnostic and treatment options. Ophthalmic Surg Lasers Imaging Retina 2019;50:11:670-674.
61. Augsburger JJ, Coats TD, Lauritzen K. Localized suprachoroidal hematomas. Ophthalmoscopic features, fluorescein angiography, and clinical course. Arch Ophthalmol 1990;108:7:968-72.