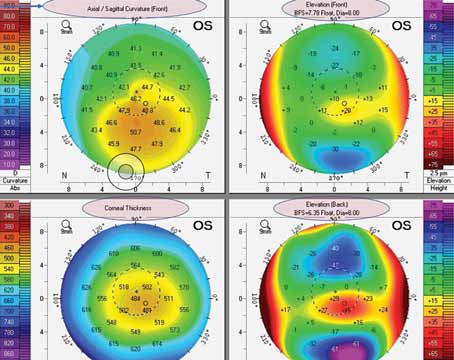
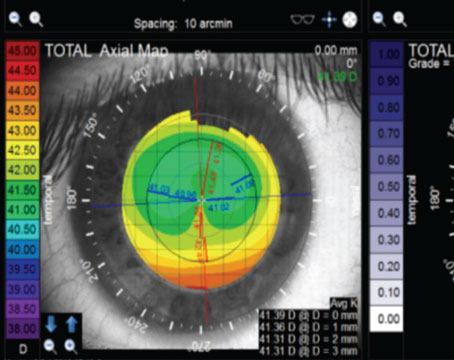
The incidence of congenital corneal opacities, including congenital glaucoma, is six per 100,000. CHED makes up a small minority.3 Although the incidence of CHED is quite low, it is more common in places with higher consanguinity. In reviews from the Middle East and India, a diagnosis of CHED accounted for 21 percent of all pediatric keratoplasties.4,5
 |
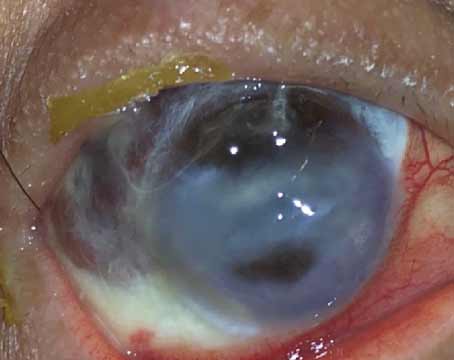
| Figure 1. A 3-year-old female with congenital hereditary endothelial dystrophy. The right eye is 1.5 months post-DSAEK without endothelial stripping. There is increased corneal clarity. The left eye has not been operated on yet. There is a mild but diffuse corneal clouding, which in this picture can best be appreciated over the pupil. |
Until recently, CHED had been classified into two variants: 1) an autosomal dominant variant that presents after 1 year of age and is progressive (CHED1); and 2) an autosomal recessive variant that presents at birth and is stationary (CHED2). Patients with CHED1 are described as being born with clear corneas and develop clouding over the first few years of life. They complain of epiphora and photophobia, but do not have nystagmus. By contrast, CHED2 patients are described as having corneal clouding from birth with accompanying nystagmus. They do not complain of epiphora or photophobia. The corneal clouding in CHED2 is generally more advanced than in CHED1, and corresponds to a worse visual acuity. Recent genetic studies have mapped CHED1 to an area on chromosome 20 that overlaps with posterior polymorphous corneal dystrophy (PPCD).8 Additionally, a review of reported cases of CHED1 found considerable overlap in clinical, histopathologic and electron microscopic examinations between CHED1 and PPCD.9 Clinically, patients with both conditions have been described as having small flakes, spots and irregular white areas throughout the stroma. A review of histopathologic and electron microscopy studies of previously diagnosed CHED1 patients reveals multiple abnormal endothelial layers and microvilli, which can also classically be seen with PPCD.10,11 Recently, the international classification of corneal dystrophies (IC3D) has been revised to eliminate CHED1 from classification, stating that, “There is no convincing published evidence to support the existence of AD CHED as a distinct entity.”12 The autosomal recessive CHED (CHED2) is now renamed CHED.
Clinical Findings
In addition to corneal clouding and thickened pachymetry, patients with CHED may have an irregular or roughened appearance of their epithelium, suggestive of edema. This can be better delineated by the instillation of fluorescein. The clouding in CHED is diffuse (limbus to limbus) and relatively uniform. While the cornea can be quite cloudy, even milky, the iris is usually visible. This is in contrast to other etiologies of congenital corneal clouding like Peter’s anomaly or a limbal dermoid, which can vary in size/location and often obscure underlying iris. Aside from the corneal abnormalities, the structure of the eye in CHED patients is usually normal.
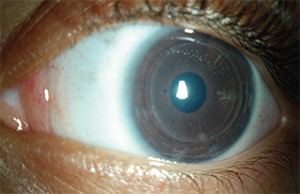 |
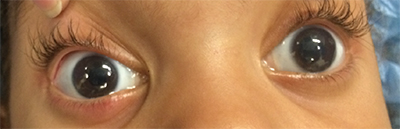
| Figure 2. A 3-year-old female with CHED, three months after DSAEK without endothelial stripping of the left eye. Her corneal clarity is significantly improved and her visual acuity improved from 20/250 to 20/80. |
Clinicians should also inquire about hearing loss, as several patients with CHED have been found to have Harboyan syndrome, also called corneal dystrophy and perceptive deafness. These patients have progressive post-lingual sensorineural hearing loss. The hearing loss typically becomes clinically apparent by 10 to 25 years old. Deficits start in the 20 to 25 dB range (mild to moderate) and occur at higher frequencies.18 More than 50 percent of cases have been associated with parental consanguinity.
Genetics
CHED has been mapped to chromosome 20 loci 20p13. The gene codes solute carrier family 4, sodium borate transporter member 11 (SLC4A11).8 Eranga Vithana and colleagues studied 10 families with CHED and found seven different mutations in the SLC4A11 gene.19 The gene encodes a sodium borate transporter. Transfection of an immortalized cell line with mutant SLC4A11 cDNAS resulted in little to no expression of transporter protein on the cell surface. The majority of pedigrees with CHED who have been screened demonstrate coding region mutations in the SLC4A11 gene.9 It is thought that the intracellular boron concentration plays a key role in the growth and terminal differentiation of neural crest cells. Therefore, loss of function of this transporter may result in abnormal corneal endothelial synthesis and differentiation.
Patients with Harboyan syndrome also have mutations in the SLC4A11 gene. The gene is expressed in the cochlea of adult mice and thought to play a role in the homeostasis of cochlear fluid and endolymph secretion.18 This may explain the concomitant corneal edema and hearing loss seen in patients with Harboyan syndrome.
Management
CHED patients have varying visual acuities depending on the degree of corneal clouding. A decision must be made whether to intervene or observe. If intervention is desired, surgery is required. The pathophysiology is a result of inadequate endothelium, and treatment therefore requires replacing the endothelium. This can be accomplished by either a full-thickness transplant (penetrating keratoplasty) or a partial-thickness transplant surgery, where only the endothelium is replaced (Descemet’s stripping endothelial keratoplasty). In all cases, patients should be carefully followed and amblyopia should be treated.
Penetrating Keratoplasty
PK in children can be challenging for a number of reasons: Children have higher rates of graft rejection than adults. Surgery is challenging because of lower scleral rigidity, increased fibrin reaction and positive vitreous pressure. Children may be difficult to examine and suture adjustments may necessitate multiple exams under anesthesia, all the while taking into account changing refractions and managing amblyopia.5
A multicenter retrospective study for PK in CHED by Debra A. Schaumberg, MPH, and colleagues found a first graft survival rate of 71 percent.20 Half of the children improved one or more Snellen lines, but only 40 percent ultimately achieved visual acuity of 20/200 or better.21 A second series from India found a 75 percent graft survival rate at four years for presumed AR CHED patients. Ali A. Al Rajhi, MD, and colleagues compared “early onset CHED,” formerly CHED2, to “delayed onset CHED,” formerly CHED1 but now considered part of the PPCD spectrum. They found better acuities and graft survival in the delayed onset group.22 Similarly, series where patients received grafts at later ages report improved visual acuity and graft survival.23,24 However, this relative success may be a function of the fact that these patients have more mild disease/PPCD.
DSEK
Given the fact that CHED is primarily a disorder of the corneal endothelium and Descemet’s membrane, a DSEK is another good surgical option. Compared to PK, DSEK offers the advantage of faster visual recovery, a relatively closed system during surgery and fewer corneal sutures, producing less corneal astigmatism and necessitating less postoperative suture adjustment/removal. One challenge of DSEK in CHED is Descemet’s scoring and removing the corneal endothelium. Visibility is often compromised due to corneal clouding, and the endothelium has been found to be more adherent than in other cases of endothelial dysfunction, like Fuchs’ dystrophy.25 A second challenge is the fact that CHED patients are often phakic and, therefore, causing a cataract is a significant risk during surgery. In adults, Janet Y.M. Tsui, MD, and colleagues found that the presence of an air bubble caused a premature cataract in 40 percent of phakic eyes.26 In a study at L.V. Prasad Eye Institute, Jatin N. Ashar, MD, and colleagues did a paired comparison, where patients with CHED underwent PK in one eye and DSEK in the contralateral eye.27 At one year, they found that all grafts were clear. In the DSEK group, the astigmatism was significantly lower and had faster stabilization of refraction. By contrast, grafts appeared overall clearer in the PK group, although final visual acuity was equivalent. Several case series have reported improved visual acuity and corneal clarity with a relatively short visual recovery in patients undergoing DSEK for CHED.28,29
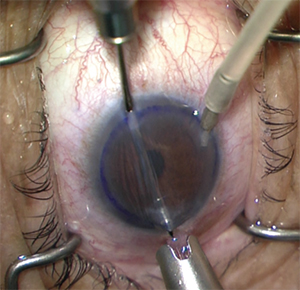 |
| Figure 3. Performing a DSAEK with a Busin glide, anterior chamber maintainer and a miotic pupil. |
Given the difficulty of Descemet’s stripping in the setting of reduced corneal clarity and adherent endothelium, several authors have completed DSEK in CHED without stripping and without removing the host endothelium. Massimo Busin, MD, and colleagues did not strip Descemet’s in patients less than 1 year old at the time of surgery because Descemet’s could not be identified. In all patients, the donor tissue ultimately attached and the cornea cleared completely by one week after surgery, although in four of the six eyes, re-bubbling was necessary.29 Jatin N. Ashar, MD, and colleagues compared patients with CHED where Descemet’s was not stripped (nDSEK) to those where it was stripped (DSEK), and found comparable final visual acuities between the two groups.30 One of the three patients with nDSEK needed to be re-bubbled compared to 0 with DSEK. Based on these findings, DSEK without stripping is a viable option for patients where there is poor visibility, who are very young and/or Descemet’s is difficult to identify and remove.
Several techniques have been described to minimize lens trauma in phakic pediatric DSEK patients. Mahmoodreza Panahi-Bazaz, MD, and colleagues describe a suture pull-through method, to minimize anterior chamber manipulation when visibility is poor.31 Additionally, topical pilocarpine was used to induce miosis. Dr. Busin and colleagues advocate moving the incision superiorly so that the pull-through method can be done over the superior iris, which protects the underlying crystalline lens.29
Prognosis
Final visual outcomes in patients with CHED vary and depend on the degree of corneal clouding, the level of amblyopia and whether or not there was a successful surgical intervention. Compared to other etiologies of congenital corneal clouding, CHED patients tend to have better surgical outcomes.5 This is likely due to the fact that, in most circumstances, CHED patients have an otherwise healthy eye. The recent advent of DSEK offers a much less invasive alternative to PK surgery. Clinicians should screen carefully for concomitant conditions like glaucoma and hearing loss. Patients and family should be counseled on the necessity of long-term follow-up and should understand the genetic nature of this disease. REVIEW
Dr. Trief is an assistant professor of ophthalmology at the Edward S. Harkness Eye Institute, Columbia University Medical Center. Dr. Ritterband is a clinical professor of ophthalmology at the Icahn School of Medicine of Mt. Sinai, and the assistant director of the cornea service at the New York Eye and Ear Infirmary of Mt. Sinai. Dr. Seedor is a clinical professor of ophthalmology at the Icahn School of Medicine, and system director of cornea and external disease at NYEEI of Mt. Sinai. Dr. Waisbren is an assistant professor of ophthalmology at Icahn. Also contributing to the article were Luna Xu, MD, and Yijie Lin, MD, MBA, ophthalmology residents at NYEEI of Mt. Sinai.
1. Maumenee AE. Congenital hereditary corneal dystrophy. Am J Ophthalmol 1960;50:1114-24.
2. Nischal KK. A new approach to the classification of neonatal corneal opacities. Curr Opin Ophthalmol 2012;23:344-54.
3. Bermejo E, Martinez-Frias ML. Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am J Med Genet 1998;75:497-504.
4. Al-Ghamdi A, Al-Rajhi A, Wagoner MD. Primary pediatric keratoplasty: Indications, graft survival, and visual outcome. J AAPOS 2007;11:41-7.
5. Vanathi M, Panda A, Vengayil S, Chaudhuri Z, Dada T. Pediatric keratoplasty. Surv Ophthalmol 2009;54:245-71.
6. Jay H. Krachmer MJM, Edward J. Holland, ed. Cornea, 3rd Edition. Philadelphia: Elsevier Mosby; 2011.
7. Kenyon KR, Antine B. The pathogenesis of congenital hereditary endothelial dystrophy of the cornea. Am J Ophthalmol 1971;72:787-95.
8. Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet 1995;4:2395-8.
9. Aldave AJ, Han J, Frausto RF. Genetics of the corneal endothelial dystrophies: An evidence-based review. Clin Genet 2013;84:109-19.
10. Kanai A, Kaufman HE. Further electron microscopic study of hereditary corneal edema. Invest Ophthalmol 1971;10:545-54.
11. Kanai A, Waltman S, Polack FM, Kaufman HE. Electron microscopic study of hereditary corneal edema. Invest Ophthalmol 1971;10:89-99.
12. Weiss JS, Moller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies--edition 2. Cornea 2015;34:117-59.
13. Khan AO, Al-Shehah A, Ghadhfan FE. High measured intraocular pressure in children with recessive congenital hereditary endothelial dystrophy. J Pediatr Ophthalmol Strabismus 2010;47:29-33.
14. Ramamurthy B, Sachdeva V, Mandal AK, Vemuganti GK, Garg P, Sangwan VS. Coexistent congenital hereditary endothelial dystrophy and congenital glaucoma. Cornea 2007;26:647-9.
15. Mullaney PB, Risco JM, Teichmann K, Millar L. Congenital hereditary endothelial dystrophy associated with glaucoma. Ophthalmology 1995;102:186-92.
16. Wilson ME. Congenital iris ectropion and a new classification for anterior segment dysgenesis. J Pediatr Ophthalmol Strabismus 1990;27:48-55.
17. Nischal KK. Congenital corneal opacities - A surgical approach to nomenclature and classification. Eye (Lond) 2007;21:1326-37.
18. Desir J, Abramowicz M. Congenital hereditary endothelial dystrophy with progressive sensorineural deafness (Harboyan syndrome). Orphanet J Rare Dis 2008;3:28.
19. Vithana EN, Morgan P, Sundaresan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet 2006;38:755-7.
20. Schaumberg DA, Moyes AL, Gomes JA, Dana MR. Corneal transplantation in young children with congenital hereditary endothelial dystrophy. Multicenter Pediatric Keratoplasty Study. Am J Ophthalmol 1999;127:373-8.
21. Pandrowala H, Bansal A, Vemuganti GK, Rao GN. Frequency, distribution, and outcome of keratoplasty for corneal dystrophies at a tertiary eye care center in South India. Cornea 2004;23:541-6.
22. al-Rajhi AA, Wagoner MD. Penetrating keratoplasty in congenital hereditary endothelial dystrophy. Ophthalmology 1997;104:956-61.
23. Sajjadi H, Javadi MA, Hemmati R, Mirdeghan A, Parvin M, Nassiri N. Results of penetrating keratoplasty in CHED. Congenital hereditary endothelial dystrophy. Cornea 1995;14:18-25.
24. Ozdemir B, Kubaloglu A, Koytak A, et al. Penetrating keratoplasty in congenital hereditary endothelial dystrophy. Cornea 2012;31:359-65.
25. Pineda R, 2nd, Jain V, Shome D, Hunter DC, Natarajan S. Descemet’s stripping endothelial keratoplasty: Is it an option for congenital hereditary endothelial dystrophy? Int Ophthalmol 2010;30:307-10.
26. Tsui JY, Goins KM, Sutphin JE, Wagoner MD. Phakic descemet stripping automated endothelial keratoplasty: Prevalence and prognostic impact of postoperative cataracts. Cornea 2011;30:291-5.
27. Ashar JN, Ramappa M, Vaddavalli PK. Paired-eye comparison of Descemet’s stripping endothelial keratoplasty and penetrating keratoplasty in children with congenital hereditary endothelial dystrophy. Br J Ophthalmol 2013;97:1247-9.
28. Ashar JN, Madhavi Latha K, Vaddavalli PK. Descemet’s stripping endothelial keratoplasty (DSEK) for children with congenital hereditary endothelial dystrophy: Surgical challenges and 1-year outcomes. Graefes Arch Clin Exp Ophthalmol 2012;250:1341-5.
29. Busin M, Beltz J, Scorcia V. Descemet-stripping automated endothelial keratoplasty for congenital hereditary endothelial dystrophy. Arch Ophthalmol 2011;129:1140-6.
30. Ashar JN, Ramappa M, Chaurasia S. Endothelial keratoplasty without Descemet’s stripping in congenital hereditary endothelial dystrophy. J AAPOS 2013;17:22-4.
31. Panahi-Bazaz M, Sharifipour F, Malekahmadi M. Modified Descemet’s Stripping Automated Endothelial Keratoplasty for Congenital Hereditary Endothelial Dystrophy. J Ophthalmic Vis Res 2014;9:522-5.