If a patient complains of blurring of vision progressing over several days accompanied by pain in the eye that worsens with eye movement, she may have optic neuritis—the most common acute optic nerve disorder in people ages 15 to 45.
The recognition of optic neuritis has recently become a hot topic for physicians—especially ophthalmologists and neurologists—for two reasons: Optic neuritis often is the first indication of the potentially debilitating neurological disease multiple sclerosis. More than 400,000 Americans suffer with MS, a leading cause of progressive disability in young adults.
Early intervention with new immunomodulatory agents can delay onset of clinically definite MS and mitigate the debilitating course of MS to improve our patients' long-term prognosis. The treatment is expensive, however, and the side effects can be onerous. This makes the accurate diagnosis of optic neuritis more important than ever in order to treat everyone at high risk of developing MS but to avoid subjecting patients at low risk for MS to unnecessary medication.
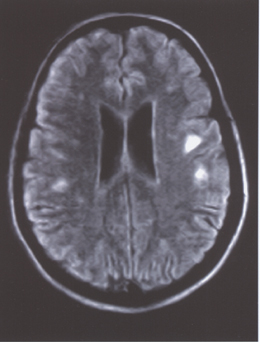 |
| This MRI of a patient with optic neuritis shows multiple hyperintense foci in the white matter, consistent with multiple sclerosis. |
Optic neuritis is an inflammation of the optic nerve, characterized by blurred vision with decreased acuity, defective color vision and visual field loss. The patient may complain that red appears gray or dark. The visual field defect reflects demyelinating axons and while classically cecocentral, can also be arcuate or altitudinal.
Constriction of the pupils provides an objective measure of the light seen by an eye. An ophthalmologist suspecting optic neuritis should always look for a relative afferent pupillary defect by swinging a light back and forth from the left eye to the right eye to the left eye, observing the decreased pupillary response in the involved eye. In only one-third of cases, examination of the fundus finds a swollen optic nerve head. In two-thirds of cases, the optic disc initially looks normal because the inflammation is retrobulbar.
Who Gets Optic Neuritis
Optic neuritis has been considered an early form of multiple sclerosis, an autoimmune disorder in which T-cells surround blood vessels, disrupt the myelin sheath that insulates the nerve axons, and may attack the nerves themselves. Most MS patients develop ON at some time in the course of their disease, and optic neuritis is the presenting symptom for about 20 percent of MS patients.
The incidence of ON varies with the population, occurring most commonly in young women of northern European descent (incidence of 6/100,000 per year). It is less common in African Americans but often leads to more severe visual loss. The percentage of optic neuritis patients who go on to develop clinically definite MS—defined as two bouts of neurologic dysfunction separated in time and space—increases with time after the initial attack. Predicting which ON patients will develop MS has engendered much discussion.
Two major longitudinal prospective studies have investigated treatments to alter the likelihood of developing MS after optic neuritis. The Optic Neuritis Treatment Trial was designed to evaluate the effect of two different steroid regimens on acute, isolated ON. The Controlled High Risk Avonex Multiple Sclerosis Prevention Study evaluated the benefit of Interferon ß-1a (INFß) therapy started after a first demyelinating event, which for half their patients was ON.
ONTT Changed the Rules
Twenty years ago, when a patient was diagnosed with ON, the physician might discuss the possibility of developing MS in the future or choose not to, since the diagnosis of MS depended on the patient having a second neurologic attack at some future time, maybe years away. Faced with a patient having an initial episode of ON, many physicians prescribed oral prednisone, some IV-steroids, and others opted to do nothing. The ONTT, begun in 1988, enrolled patients with a first bout of ON and evaluated the role of corticosteroids in recovery from acute ON. The study changed the course of treatment for ON and MS.
| Typical Case of Demyelinating Optic Neuritis | ||
| History A 32-year-old woman developed left eye pain on eye movement for two days and a gradual clouding of vision in her left eye. Initially she thought her glasses were dirty. The clouding progressed over five to six days until the patient was no longer able to count fingers through that eye. The right eye seemed normal. She was in good health with no prior medical history and took no medications. She admitted to feeling fatigued after drinking hot beverages. | ||
| Exam | ||
| Right Eye | Left Eye | |
| Visual Acuity | 20/20 | Hand Motion |
| AOHRR Color Plates | 6/6 | 0/6 |
| Visual fields | Full | Cecocentral defect |
| Pupils | 0.9 log units | Left relative afferent pupillary defect |
| Motility | Full | Full |
| Fundus | Nl | Nl, pink, sharp margins, c/d 0.3 |
Regardless of whether they received placebo or either steroid regimen, most patients with ON recovered good vision, including acuity, color, contrast sensitivity and visual field. Intravenous Solumedrol hastened recovery from ON but didn't improve the eventual visual outcome at one year. The treatment groups differed in their likelihood of having a recurrence of ON.
Patients who received only oral prednisone were twice as likely to have another attack of ON in either eye than patients who received Solumedrol or placebo.
In the two years following the initial attack of ON, new attacks of ON developed in 30 percent of the patients treated with oral prednisone, compared with 14 percent in the Solumedrol group and 16 percent in the placebo group.
Thus, oral prednisone not only failed to improve recovery from ON, but increased the risk of having another attack of ON in one eye or the other within two years after treatment compared with the IV Solumedrol-treated patients, as well as with the control group. These findings resulted in oral prednisone at that dose becoming contraindicated in the treatment of ON.
Of interest, the patients who received IV Solumedrol treatment developed MS at a lower rate over the first two-year period than those treated with prednisone or placebo. After two years, definite MS had developed in only 7.5 percent of Solumedrol-treated patients compared with 13 percent of oral prednisone-treated patients and 17 percent of those who received placebo.
The benefit of Solumedrol seemed to wane after two years. Nevertheless, IV Solumedrol became the treatment of choice for ON patients, especially those who need to recover vision more quickly.
The study group that established the ONTT has been following a cohort of patients who had ON between 1988 and 1991. Recently, they published their 10-12 year follow-up data showing that the overall risk of a diagnosis of clinically definite MS after ON was 30 percent at five years and close to 40 percent at 10 years. Rates were worse, however, for a high-risk group composed of those patients who had one or more 3-mm white matter plaques on MRI at the time of the initial bout of ON. Patients with demyelinating lesions on MRI had a 56-percent risk of developing clinically definite MS within 10 years after a single bout of ON.
On the other hand, those patients who had no MRI lesions at the time of their ON had a MS risk of only 18 percent. The absence of certain clinical features typical of demyelinating ON, also lowered the risk of MS.
Importance of MRI
Although neuroimaging did not contribute to making the initial diagnosis of ON, the presence or absence of a demyelinating lesion on MRI was correlated with the risk of developing clinically definite MS. At first, the number of lesions on MRI seemed to correlate with the risk of clinically definite MS. In their recent 10-12 year follow-up, however, the ONTT reported that the presence of even one demyelinated plaque (i.e. a single 3-mm diameter white matter lesion visible on the brain MRI) placed the patient at a higher risk for MS than if lesions were absent on MRI. The presence of more lesions did not increase the risk further. In those patients with no MRI lesions, the rate at which definite MS developed was so low that no therapeutic effect could be measured. For prognostic purposes, an MRI should be done at the time of the first demyelinating event.
MS plaques show up on MRI as hypointense lesions on T1-weighted images, hyperintense lesions on T2 and enhancing lesions after gadolinium.
Hyperintense T2 lesions are commonly counted to assess the extent of disease. T2-weighted images can be modified and made more sensitive with proton density sequences or by FLAIR, or fluid attenuated inversion recovery sequences. FLAIR is likely to show more lesions and indicate active disease.
Despite the dogma that demyelinating MRI lesions are tantamount to a diagnosis of MS, 10 years after the initial presentation with ON with a lesion on MRI, only 60 percent of patients have gone on to clinically definite MS; 40 percent have remained well. The low-risk group is characterized by the absence of MRI lesions and absence of a history of pain. Men are at lower risk than women.
Other details of the ophthalmic exam can help to predict which patients are at risk for a recurrence and which are in a low-risk group and would do well without treatment. Specifically, severe optic disc swelling with hemorrhages, exudates, and a macular star are unusual for MS. So if a patient comes in with ON but has no lesions on MRI, the study suggests that he will recover his vision without therapy and will continue to do well and not develop clinically definite MS even without disease-altering drugs.
CHAMPS
Unfortunately, MS may cause degeneration in the brain even when it does not produce acute attacks. Neurologists have tried to quantify the "burden of disease" by computing the volume of demyelinating lesions in the brain, measured by hyperintense lesions on T2. Decreases in lesion load have been documented after treatment with the interferons, Avonex, Rebif or Betaseron, and with glatiramer acetate (Copaxone).
The Controlled High Risk Avonex Multiple Sclerosis Prevention Study provided evidence that early treatment with Interferon ß-1A (Avonex) at the time of the first clinical demyelinating episode seems to delay the development of clinically definite MS and prevent disability. CHAMPS enrolled patients with an isolated demyelinating neurological event: optic neuritis, cerebellar-brain stem syndrome or spinal-cord syndrome. Patients must have had two or more MRI lesions that put them at high risk for MS. Patients were assigned randomly to receive placebo or intramuscular Avonex 30 mg once-a-week.
Upon enrollment, patients received a course of IV-steroid treatment with oral taper, similar to the ONTT protocol, and then began weekly intramuscular injections of Avonex or placebo. Compared to placebo, Avonex significantly delayed the onset of clinically definite MS. The effect was so robust that the three-year trial was stopped early.
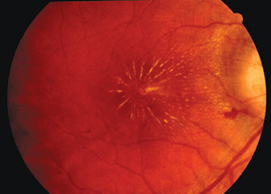 |
| Fundus photo of a patient with neuroretinitis. The presence of disc swelling, hemorrhages and macular star are atypical for demyelinating optic neuritis and put the patient at lower risk for multiple sclerosis. |
Over the course of the trial clinically definite MS developed in 50 percent of the placebo-treated patients but only 35 percent of patients in the Avonex group, a 44-percent reduction. More impressive were the MRI findings indicating that the Avonex group had a slower increase in the volume of brain lesions, as well as 57-percent fewer MRI lesions. Similar protective results have been reported by the Early Treatment of MS using Rebif study. Such data showing delay of clinically definite MS and decreased burden of disease strongly encourage early intervention in relapsing demyelinating disease.
Early Intervention
Prompt diagnosis of ON and early treatment with INFß results in deferral of clinically definite MS, fewer demyelinating events and a smaller burden of disease as measured by the volume of plaques seen in the brain on MRI. It is hoped that early intervention with immunomodulating agents will decrease physical disability and cognitive decline from this chronic progressive disease. Early intervention dictates that every patient with ON undergo MRI and those with a demyelinating brain lesion on MRI be treated with IV Solumedrol and referred to a neurologist for treatment with IM Avonex (or Rebif, Betaseron or Copaxone). A patient who does not meet the CHAMPS entry requirements should not be treated, but should be followed.
| Diseases Masquerading As Optic Neuritis |
| Ophthalmologists have to be more careful than ever not to misdiagnose retinal syndromes and other optic-nerve diseases as demyelinating optic neuritis associated with multiple sclerosis. Here are the most common conditions that can mimic ON: |
| Other Optic Neuropathies |
| • Anterior ischemic optic neuropathy |
| • Autoimmune optic neuropathy |
| • Toxic or nutritional optic neuropathy |
| • Leber's hereditary optic neuropathy |
| • Compressive optic neuropathy |
| • Glaucoma |
| • Traumatic optic neuropathy |
| • Perineuritis |
| Macular/Retinal Diseases |
| • Neuroretinitis |
| • Central serous choroidopathy |
| • Flat retinal detachment |
| • Acute zonal outer retinopathy |
| • Acute macular neuroretinitis |
Another 30-year-old woman was treated according to the ONTT with IV Solumedrol in 1993 and returned with ON in the same eye nine years later. Her MRI showed a retrobulbar enhancing lesion. She admitted to Uhthoff symptom—generalized weakness and blurred vision when she was overheated—strongly suggestive of MS. She requested Solumedrol to hasten her recovery and consulted a neurologist but would not start Avonex. She has remained well for two years since then.
A third woman, 35, had ON with several lesions on MRI, and was treated with IV Solumedrol. She consulted a neurologist who found a normal exam, but recommended Avonex. She decided to defer treatment and has been asymptomatic for two years. Her doctors are worriedly anticipating another attack, but the patient congratulates herself on avoiding the lifestyle change necessitated by taking INFß, involving regular IM or subcutaneous injections with side effects including not only hematomas at the injection site, but also headache, flu-like symptoms, hair loss and nausea. Fortunately, the immunomodulating agents can always be started in a patient with established MS with good effect.
Nailing the Diagnosis
The role of the ophthalmologist or neuro-ophthalmologist is to make the correct diagnosis of ON, noting typical and atypical features of the history and exam. It is important to discriminate between ON associated with MS and other conditions that mimic or may be mistaken for ON (See box above), such as ischemic optic neuropathy, sarcoid, syphilis and neuroretinitis.
The examination of ON should include:
• History. Ask detailed questions about the loss of vision and whether it is accompanied by pain. What distinguishes ON from other neuropathies is pain upon eye movement, accompanying gradual vision loss that progresses over three to 10 days. Sudden vision loss that occurs on awakening is more likely caused by ischemia than by ON. Gradual vision loss progressing for a month or more is more likely to describe a compressive lesion.
Ask the patient whether she has ever experienced symptoms of MS such as fatigue, Uhthoff symptom, L'Hermitte's sign (tingling at the base of the spine when flexing the neck), weakness, pain, tingling or numbness in the arms and legs, incoordination and bowel and urination difficulties.
Question the patient about symptoms of other autoimmune disease or other causes of ON such as sarcoidosis, lupus erythematosus or recent infections. If the history is negative, blood tests need not be ordered.
• Eye exam. Measure visual acuity and color vision. Record the visual field on a Humphrey 24-2 threshold test including foveal sensitivity. Check for the presence of relative afferent pupillary defect and quantify the defect with neutral density filters, if possible. This will enable determination at a follow-up visit of whether the patient's vision has improved. You'll want to check for ocular motility abnormalities common in MS, such as internuclear ophthalmoplegia or nystagmus.
• Schedule MRI. Have your patient undergo MRI of brain and orbits with and without gadolinium. Look for black holes on T1, hyperintense lesions on T2, especially with proton density and FLAIR, and gadolinium-enhanced lesions.
Follow-Up
If the neuro-ophthalmic patient has one or more lesions on her MRI, she has a much higher risk for developing MS than someone who has no lesions. A three-day course of Solumedrol can be administered to speed recovery and to decrease the risk of clinically definite MS over the next two years.
The protocol for Solumedrol treatment has been variously modified without scientific verification of efficacy. Without knowledge of the critical parameters of steroid dosage, many practitioners have tinkered with the Solumedrol protocol used by the ONTT. Specifically, since IV steroids are now usually administered on an outpatient basis, Solumedrol 1 g per day for three or four days is commonly given instead of divided doses, and the oral taper is frequently omitted. Controlled trials have not been done to establish the efficacy of these modified protocols. The reasons for the poor outcome of oral prednisone-treated patients in the ONTT remain hypothetical. Oral prednisone, however, is still contraindicated.
Partnership with Neurology
The patient with typical, isolated, monosymptomatic ON should be referred promptly to a neurologist who can begin immunomodulatory medication. The results of the neuro-ophthalmic exam should accompany the patient to the neurologic consultation. The neurologist relies on the ophthalmologist to accurately diagnose ON and to distinguish demyelinating ON from other optic neuropathies or retinal conditions that have similar symptoms.
The neurologist will want neuro-ophthalmic or ophthalmic follow-up including an exam six to 12 weeks after onset to assess the degree of recovery from the acute ON. Periodic examinations after that will assess the stability or progression of any visual field defects.
Physicians have reason to be more hopeful and optimistic about the prognosis for MS presenting as ON and the long-term prognosis of MS. We have new information about the levels of risk for developing MS after neurologic events, and we have immune-modulating therapies. Ophthalmologists should sharpen diagnostic and decision-making skills to discern which patients will benefit most from these therapies.
Dr. Winterkorn is a neuro-ophthalmologist in private practice and a clinical professor of neurology and neuroscience and of ophthalmology at the New York-Presbyterian Hospital-Weill Medical College of Cornell University.

1. Optic Neuritis Study Group. The clinical profile of acute optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol 1991;109:1673-1678.
2. Beck RW, Cleary PA, Anderson MA, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med 1992;326:581-588.
3. Beck RW, Cleary PA, Trobe JD, et al. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. N Engl J Med 1993;329:1764-1769.
4. Beck RW, Trobe JD. The Optic Neuritis Treatment Trial: putting the results in perspective. J Neuro-ophthalmology 1995;15:3:131-135.
5. Five year risk of multiple sclerosis after optic neuritis: experience of the ONTT Neuritis Study Group. Neurology 1997;49:1404-1413.
6. Jacobs LD, Beck RW, Simon JH, Kinkel RP, et al. Intramuscular Interferon ß-1A Therapy Initiated During a First Demyelinating Event in Multiple Sclerosis. The CHAMPS Study Group. N Engl J Med 2000;343:3:898-904.
7. Beck RW, Trobe JD, Moke PS, et al. High and Low Risk Profiles for the Development of MS within 10 years after Optic neuritis. Arch Ophthalmol 2003; 121:944-949.


