Workup, Diagnosis and Treatment
At the time of diagnosis, optical coherence tomography of the macula was normal bilaterally without evidence of cystoid macula edema or epiretinal membrane. B-scan ultrasonography OD revealed a highly reflective retinal mass 2.5 mm in thickness. Fluorescein angiography disclosed a dilated and tortuous artery and vein as well as staining and leakage of the mass.
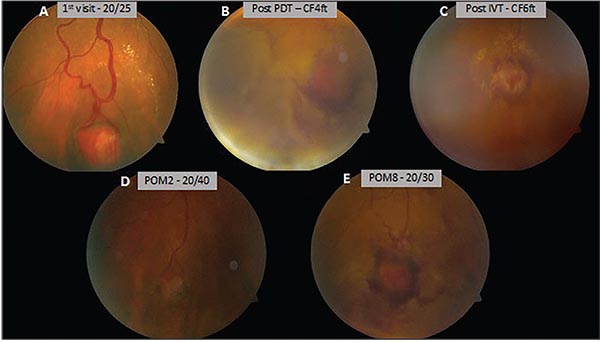 |
| Figure 2. Peripheral fundus photographs of the right eye, documenting the retinal hemangioblastoma at the time of diagnosis (A), after initial photodynamic therapy (B), following intravitreal preservative-free triamcinolone acetonide (C), and at postoperative months two (D) and eight (E). |
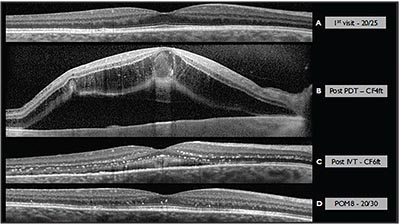 |
| Figure 3. OCT of the macula documenting the grossly normal macular contour at time of diagnosis (A), as well as the presence of cystoid macular edema after initial photodynamic therapy (B). Following intravitreal injection of preservative-free triamcinolone acetonide (C), and at postoperative month eight (D), hard exudates remained in the outer and inner nuclear layers, in addition to subretinal debris and shallow subfoveal fluid. |
Therapeutic options were discussed, including observation (given the absence of macular involvement), anti-VEGF intravitreal injection, photodynamic therapy, and laser photocoagulation to the hemangioblastoma. The patient elected PDT, which proceeded using standard settings (50 J/cm2 with a 689-nm laser for 83 seconds). Following PDT, the patient noted gradually reduced vision to CF at 4 feet and was found to have an exudative retinal detachment adjacent to the tumor and cystoid macular edema. She was treated with intravitreal preservative-free triamcinolone acetonide 4 mg/0.1 cc. Slowly, the tumor resolved over several months after PDT, with the subretinal fluid and cystoid macular edema also resolving. Visual acuity returned to 20/30 in the affected eye. Figures 2 and 3 document the initial presentation and the course of the disease over time in this case.
Discussion
Retinal hemangioblastoma (RH, also known as retinal capillary hemangioma) is classically described as a red-orange, well-defined retinal mass with prominent dilated feeding vessels. These tumors are typically classified by their location (peripheral vs. juxtapapillary), morphology (endophytic vs. exophytic vs. sessile), local effects on the retina (exudation or vitreoretinal traction), and the presence or absence of the VHL mutation. Most RH are found in the peripheral retina with a proclivity for the superotemporal and inferotemporal quadrants. The natural history of RH is variable, with the potential for progression, regression or clinical stability.1
RH pathology is notable for vascular channels lined by foamy stromal cells, which are postulated to be neoplastic in light of their loss of heterozygosity of the VHL gene and upregulation of VEGF.1 Researchers at the National Institutes of Health conducted a prospective consecutive case series documenting 335 patients with RH in the context of VHL. Fifty-eight percent of patients had bilateral RH, and 85 percent of RH masses were located in the peripheral retina.2 A 2001 retrospective consecutive case series of 68 patients diagnosed with RH at the Ocular Oncology Service at Wills Eye Hospital revealed that the median age of diagnosis with associated VHL was 18 years younger than for patients with sporadic RH.3 In the study, sporadic RH tended to affect middle-aged individuals (approximately 36 to 48 years old) with unilateral, solitary tumors.1 Approximately 46 percent of solitary RH are associated with VHL.4
VHL is an inherited autosomal dominant cancer predisposition syndrome carried on chromosome 3 (3p25-26).1 This condition is caused by a germline mutation in the VHL tumor suppressor gene and affects approximately 1 in 40,000 births; approximately 20 percent are de novo mutations.1 Mounting evidence supports the classical “twohit” hypothesis in which patients have an initial germline mutation resulting in the loss of a tumor suppressor such as VHL in all cells, followed by local loss of the second (and sole functioning) allele, which results in complete loss of tumor suppression.3
Common manifestations of VHL include RH, CNS hemangioblastoma, pheochromocytoma, renal cell carcinoma (RCC), and cysts of the kidneys and pancreas.1,5 RH is often the presenting feature, but rarer ophthalmic findings associated with VHL have been reported, including twin vessels (paired retinal arteriole and venule, separated by less than one venule diameter) and vision loss secondary to CNS hemangiomas.1,6 Average life expectancy of patients with VHL is between 40 and 52 years of life, with the most common causes of death being RCC and CNS hemangioblastoma.
A diagnosis of VHL reflects both clinical and genetic findings. In patients without a known family history of VHL, a diagnosis may be made based on the presence of two of more of the following: two or more hemangioblastomas affecting the retina, spine or brain; a single other visceral hemangioma; RCC; pheochromocytoma; or, more rarely, endolymphatic sac tumors; papillary cystadenomas; or pancreatic neuroendocrine tumors. Alternatively, patients with a known family history only need one of more of the following features: retinal angioma; CNS hemangioblastoma; pheochromocytoma; RCC; or multiple cysts of the kidney or pancreas.7 VHL genetic testing is positive in more than 95 percent of patients who meet the clinical diagnostic criteria for VHL.8
VHL can be classified based on the absence or presence of CNS hemangioblastoma, pheochromocytoma and RCC. Type 1A is characterized by CNS hemangioblastoma and RCC. Type 1B is marked by CNS hemangioblastoma in the absence of RCC. Type 2 is characterized by the presence of pheochromocytoma and further subclassified based on the presence of CNS hemangioblastoma (Type 2A), CNS hemangioblastoma and RCC (Type 2B) or pheochromocytoma only (Type 2C).9
Upon diagnosis, patients are rigorously screened for manifestations of the VHL syndrome, including CNS hemangioblastomas, pheochromocytomas and RCC.10 Of note, an annual ophthalmic exam is recommended for VHL patients of all ages.11 The Wills Eye Hospital Ocular Oncology Service takes a multidisciplinary approach to monitoring patients with VHL. All patients undergo systemic evaluation and genetic testing at the University of Pennsylvania’s VHL clinic and undergo ophthalmic screening annually at Wills.
Treatment options for RH include close observation, laser photocoagulation surrounding the tumor, PDT, cryotherapy or plaque radiotherapy directed at the tumor. The preferred treatment is determined by the size of the RH. Small RH (<3 mm) respond well to laser photocoagulation or cryotherapy, while cryotherapy or PDT is more appropriate for medium (3 to 6 mm) RH. In our experience, larger RH (>6 mm) require PDT, plaque radiotherapy or internal resection.
Our patient elected to undergo photodynamic therapy and declined anti-VEGF. Initial treatment with PDT was complicated by an adjacent exudative retinal detachment and reduction of vision from 20/25 to counting fingers. In retrospect, this patient might have benefited from additional anti-VEGF at the time of the initial PDT administration. She was subsequently treated with intravitreal preservativefree triamcinolone acetonide to address macular edema and the exudative retinal detachment, both of which successfully responded over several months with return of vision.
In summary, our patient had a solitary peripheral retinal hemangioblastoma and negative genetic testing for VHL. She responded well to PDT with slow tumor involution over eight months.
1. Singh AD, Shields CL, Shields JA. von Hippel-Lindau disease. Surv Ophthalmol 2001;46:11742.
2. Wong WT, Agron E, Coleman HR, et al. Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with von Hippel-Lindau disease. Ophthalmology 2008;115:181-8.
3. Singh AD, Nouri M, Shields CL, et al. Retinal capillary hemangioma: A comparison of sporadic cases and cases associated with von Hippel-Lindau disease. Ophthalmology 2001;108:1907-11.
4. Singh A, Shields J, Shields C. Solitary retinal capillary hemangioma: Hereditary (von HippelLindau disease) or nonhereditary? Arch Ophthalmol 2001;119:232-4.
5. Glasker S, Neumann HPH, Koch CA, et al. Von Hippel-Lindau Disease. In: De Groot LJ, Chrousos G, Dungan K, et al., eds. Endotext (online text). South Dartmouth, Mass.:2000.
6. Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med 1990;77:1151-63.
7. van Leeuwaarde RS, Ahmad S, Links TP, et al. von Hippel-Lindau Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews (online text). Seattle:1993.
8. Binderup MLM, Stendell AS, Galanakis M, et al. Retinal hemangioblastoma: Prevalence, incidence and frequency of underlying von Hippel-Lindau disease. Br J Ophthalmol 2018;102:942-7. 9. Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet 2011;19:617-23.
10. Shields C, Shields J. Retinal tumors: Understanding clinical features, OCT morphology, and therapy. J Vitreoretinal Dis 2017;1:10-23.
11. Varshney N, Kebede AA, Owusu-Dapaah H, et al. A review of von Hippel-Lindau syndrome. J Kidney Cancer VHL 2017;4:20-9.


