APMPPE
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was first described by J. Donald M. Gass, MD, in 1968.1 It typically presents in young adults with bilateral vision loss and may be preceded by a viral illness. Presenting symptoms may include photopsia, decreased vision, paracentral scotoma or metamorphopsia. There is no gender or ethnic predilection. On fundus examination, APMPPE is characterized by randomly scattered, flat multifocal creamy white or yellow plaques at the level of the retinal pigment epithelium with indistinct margins. Plaques are typically located in the macula but may also involve the peripheral quadrants (See Figure 1A). New lesions can develop, and as such, lesions of differing age may be present at any given time.
On fluorescein angiography, active lesions demonstrate early hypofluorescence (See Figure 1B) and late staining (See Figure 1C). Inactive lesions show hyperfluorescence corresponding to window defects from retinal pigment epithelium atrophy. Indocyanine green angiography shows hypofluorescence of active and healed lesions.
|
Fundus autofluorescence in the acute phase shows hypoautofluorescence of lesions. This hypoautofluorescence may be due to a blocking effect by the outer retinal lesions versus RPE edema or direct RPE damage with decreased lipofuscin production. After resolution, lesions may demonstrate hyperautofluorescence due to deposition of lipofuscin or altered metabolism of affected RPE cells (See Figure 1E),6 but often the hypoautofluorescence persists. Further histological studies are necessary to determine the true pathogenesis and whether the primary site of damage is the outer retina and the RPE versus choroidal hypoperfusion leading to RPE and outer retinal damage.
Active plaques resolve spontaneously over a course of approximately two to four weeks, often with mottling of the RPE but without atrophy of the choroid. Visual prognosis is usually excellent, with most patients recovering their baseline visual acuity, although prognosis is less favorable when there is foveal involvement. There are uncommon cases of severe vision loss from significant RPE alterations in the fovea or due to complications from choroidal neovascularization. Recurrences are rare.
In most cases, APMPPE lesions can be observed and will resolve without any intervention. However, several reports have linked APMPPE to central nervous system vasculitis, with manifestations ranging from headaches to venous sinus thrombosis.7 This combination of APMPPE with neurologic manifestations occurs more frequently in males and very rarely can have dire consequences, including death. Treatment recommendations in such cases include IV corticosteroids followed by a slow oral taper in combination with an immunosuppressant.
Serpiginous Choroiditis
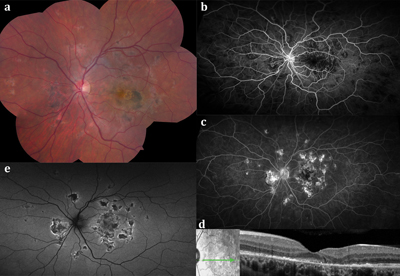
In contrast to the typically short-lived course of APMPPE, serpiginous choroiditis is characterized by bilateral, progressive and often recurrent inflammation of the choroid and outer retina.8 It usually presents in healthy, white, middle-aged adults, with a slight male predilection.9Although typically a bilateral disease, patients often present with unilateral activity and decrease in vision once the fovea is affected. Other common symptoms include small scotomas or metamorphopsia.
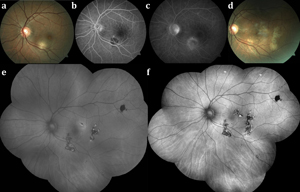
On examination, the anterior chamber is generally quiet, whereas up to 50 percent of patients may exhibit fine cells in the vitreous. Peripapillary gray-white or yellowish serpentine lesions that extend centrifugally are the classic finding by ophthalmoscopy (See Figures 2A and 2B). Recurrences are contiguous with previous lesions and often assume a pseudopodal pattern. Chorioretinal atrophy in a jigsaw-puzzle configuration may ensue (See Figure 2A). Due to the gradual extension of these geographic (serpiginous) zones of chorioretinal and RPE infiltration and then atrophy, serpiginous choroiditis has previously been referred to as “geographic helicoid peripapillary choroidopathy”10 and “geographic choroidopathy,”11 among other nomenclature.
On FA, acute serpiginous lesions demonstrate early hypofluorescence (See Figure 2C) likely due to both choriocapillaris nonperfusion and blockage from RPE and outer retinal edema. Progressive hyperfluorescence at lesion margins may represent intact choriocapillaris. Over time, there is staining of the acute lesions (See Figure 2D). ICGA shows early and late hypofluorescence of lesions, which appear larger than the corresponding lesions on examination or FA. Recurrences are common at the edges of prior atrophic scars and can occur after long periods of quiescence (even after several years). In the majority of patients the disease recurs, often several times.12
SD-OCT shows hyperreflectivity of the outer retina13 and RPE with minimal distortion of the inner retinal layers.14 Upon healing, lesions demonstrate loss of the RPE, photoreceptor outer segments, and EZ band with choroidal hyperreflectivity.14
FAF can be used to follow the course and demarcate the disease.15,16 Active inflammation appears hyperautofluorescent, while older inactive lesions appear hypoautofluorescent. A hypoautofluorescent halo that surrounds all edges of active hyperautofluorescent lesions serves as a transitional stage between active and inactive inflammation. Inactive inflammation shows dark, hypoautofluorescent lesions with sharp borders without any hyperautofluorescence at lesion edges.
With involvement of the macula, central vision is often significantly compromised. An important complication of serpiginous choroiditis is the development of choroidal neovascularization, reported to occur in about 13 to 20 percent of cases.17,18 Other complications described include cystoid macular edema, pigment epithelial detachments and branch retinal vein occlusions.19,20
|
Macular Serpiginous Choroiditis
A variant of serpiginous choroiditis termed “macular serpiginous choroiditis” was first described by Robert A. Hardy, MD, and Howard Schatz, MD, in 198710 and shares a number of features similar to classic peripapillary serpiginous choroiditis. However, in the latter entity, lesions begin around the nerve and recur centrifugally toward the macula, whereas macular serpiginous lesions commence in the macula and recur in a pseudopodal pattern centripetally towards the nerve.22 FA and ICGA findings are essentially the same and treatment strategies are similar.23 Because of its onset in the central macular region, visual acuity deteriorates early on and permanent vision loss is more profound and difficult to treat. Macular serpiginous choroiditis can also be complicated by CNV, further worsening the visual prognosis.22
Tubercular Serpiginoid Choroiditis
Included in the differential diagnosis of the placoid entities, particularly in areas of endemic tuberculosis, such as India, is tubercular serpiginoid choroiditis. It presents in either of two ways: discrete, noncontiguous multifocal choroiditis that later becomes diffuse and contiguous with an active advancing edge resembling classic serpiginous choroiditis (See Figure 3); or as a diffuse plaque-like choroiditis with amoeboid spread.24,25 Aqueous and vitreous humor aspirates from two reported cases were positive for Mycobacterium tuberculosis by polymerase-chain reaction analysis.26
In a retrospective study by Reema Bansal, MD, and colleagues, of 105 patients with confirmed TB by positive tuberculin skin testing or QuantiFERON-TB gold testing, more than 70 percent of cases were male, with a mean age of 33 years. The majority (80 percent) of patients presented with vitritis and over 60 percent had bilateral disease. Lesions were present in both the posterior pole and the periphery. In contrast to classic serpiginous choroiditis, lesions were usually not contiguous with the optic disc (>85 percent). Patients treated with antitubercular therapy (four-drug regimen) and oral corticosteroids demonstrated good responses. The addition of antitubercular treatment reduced recurrences whereas those treated with oral steroids alone had a 75 percent recurrence rate. Lesions were followed using autofluorescence to monitor response to therapy.24 Active lesions showed ill-defined hyperautofluorescence with an amorphous appearance. In the early phase of healing, a thin surrounding rim of hypoautofluorescence with a central stippled pattern developed; on complete resolution, lesions were uniformly hypoautofluorescent (See Figure 3F). With treatment, the fovea was spared in 75 percent of patients, including those with macular involvement. Up to 3.5 percent of patients developed CNV.
Interestingly, 14 percent of patients were reported to have progression of disease after initiation of antitubercular therapy.24,25 Some have proposed that tubercular serpiginoid choroiditis is an immune-mediated hypersensitivity reaction to the acid-fast bacilli sequestrated in the RPE and that dying bacilli release proteins that can paradoxically aggravate the immune-response and initially worsen the choroiditis, a type of Jarisch-Herxheimer reaction.24,25 An alternative possibility is a delayed effect of antitubercular therapy with worsening of the underlying disease by the corticosteroids. However, this subset of patients responded to increased oral corticosteroids or the addition of immunosuppressive therapy, supporting the former hypothesis. Thus, identifying the tubercular variant of serpiginous choroiditis is critical for initiation of appropriate antibiotics in combination with oral steroid therapy and one should not terminate antitubercular treatment even in the setting of initial paradoxical worsening.
Relentless Placoid Chorioretinitis
Relentless placoid chorioretinitis (RPC) is an entity that has features resembling both APMPPE and serpiginous choroiditis, but with an atypical retinal distribution and clinical time course.27 The hallmark of this disease is the development of numerous new lesions during the disease course, which may span up to two years, eventually resulting in 50 to hundreds of lesions. Peripheral lesions can precede or occur simultaneously with posterior involvement and recurrences do not necessarily initiate at the edges of prior lesions (as in serpiginous). Because of its shared features with APMPPE and serpiginous choroiditis, this entity was termed “ampiginous” by Robert B. Nussenblatt, MD.9,28
RPC presents in young patients, usually with a sudden decrease in vision and/or metamorphopsia with no history of a viral prodrome. An anterior chamber reaction or vitritis may be present.14,29 Patients exhibit small, white, placoid lesions at the level of the RPE both anterior and posterior to the equator. Some of these lesions develop pigmented chorioretinal atrophy within weeks while others have a drawn out course of persistent activity or new growth. Angiographic findings are similar to those of APMPPE and serpiginous choroidopathy.27,28,30 Only one case of RPC with OCT imaging31 and a different case with FAF imaging16 have been reported. On time-domain OCT, the active stage of the disease demonstrated hyperreflectivity of the inner and outer retinal layers with subfoveal fluid accumulation and a pigment epithelial detachment. With quiescence, normal foveal anatomy was reestablished.31 FAF revealed extensive confluent areas of marked hypoautofluorescence corresponding to areas of chorioretinal atrophy.16
Vision can decrease significantly in RPC, especially with foveal involvement. However, if treated with prolonged systemic corticosteroids, most patients can recover most of their prior vision.27 In the absence of treatment with systemic steroids, final visual acuity tends to be compromised with prolongation of disease course.28 Complications of RPC include CNV, subretinal fibrosis, subretinal fluid and epiretinal membrane formation.
Persistent Placoid Maculopathy
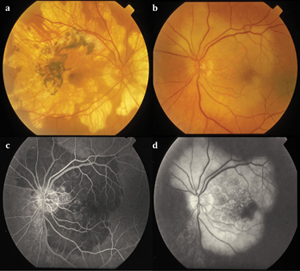
Persistent placoid maculopathy (PPM) was first described by Pamela Golchet, MD, and colleagues in 2006.32,33 It resembles macular serpiginous choroiditis in its early involvement of the macula and predominantly affects middle-aged Caucasian males. Unlike in macular serpiginous choroiditis, patients with PPM initially present with only mild visual symptoms.
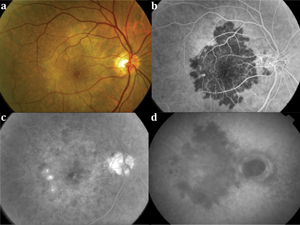
Anterior chamber inflammation or vitritis is typically absent.32,33 On ophthalmoscopy, jigsaw-pattern, whitish placoid lesions centered around the fovea that are not contiguous with the optic disc (See Figure 4A) are noted. Unlike serpiginous or macular serpiginous choroiditis, PPM lesions have a longer time course and gradually fade over months to years without the development of new lesions. FA demonstrates early hypofluorescent plaques that partially fill in late phases of the study (See Figures 4B and 4C). ICGA shows hypofluorescent plaques throughout the study (See Figure 4D). SD-OCT shows retinal thinning with photoreceptor and RPE disruption. Once the disease is stable, SD-OCT shows well-defined areas of photoreceptor loss, collapse of the outer plexiform layer and thinning of the choriocapillaris.34 In the presence of CNV, intraretinal fluid or neurosensory detachment may be present.35 FAF shows a speckled pattern of hyper- and hypo-autofluorescence in the macula.34
|
A summary of the clinical and angiographic characteristics of the spectrum of placoid white spot syndromes is provided in Table 1. In 2002, Nadia Bouchenaki, MD, and colleagues described these disorders with characteristic hypofluorescence on ICGA as indicating choriocapillaris nonperfusion and classified them as inflammatory choriocapillaropathies.30 With the aid of newer spectral-domain OCT studies, it appears there is also outer retinal involvement, and exactly where the site of primary insult resides still remains unclear. Future studies looking at enhanced depth imaging of the choroid may provide greater insight and understanding of the pathogenesis of placoid WSS. The etiology of WSS remains unknown, although the increased prevalence of systemic autoimmunity in patients with WSS and their relatives suggests WSS occurs in families with a genetic predisposition toward autoimmunity.36
Recognizing the different entities among the spectrum of the white-spot syndromes is important for the general ophthalmologist and especially for the retina specialist. Among the placoid WSS, APMPPE is the most common and benign with a viral prodrome and multiple creamy lesions that often fade within a few weeks as visual acuity recovers. Visual prognosis is more ominous in cases of serpiginous choroiditis, which endangers the fovea and carries a high risk of recurrences. The majority of patients with this particular placoid lesion permanently lose central vision in at least one eye. Relentless placoid chorioretinitis presents with numerous smaller lesions both anterior and posterior to the equator, with a prolonged clinical course and frequent relapses over months to years. With treatment, there is often only minimal sustained vision loss. Finally, persistent placoid maculopathy presents as a placoid lesion in the macula that persists for a time course of months to years; although there is only a mild decrease in visual acuity, it carries a high risk of choroidal neovascularization that may result in significant loss of central vision. Obtaining imaging studies including fundus photos, FA, ICG angiography, SD-OCT and FAF is critical to establishing the correct diagnosis and guiding management. Close clinical follow-up and providing timely treatment when indicated are paramount in preventing or at least minimizing permanent vision loss. REVIEW
Dr. George is in his final year of residency at the Jules Stein Eye Institute, UCLA, Los Angeles. Contact him at: meenageorge@gmail.com. Dr. Sarraf is a clinical professor of ophthalmology in the Retinal Disorders and Ophthalmic Genetics Division at Jules Stein. Contact him at dsarraf@ucla.edu or (310) 794-9921. Dr. Golchet practices at Southern California Permanente Medical Group in Woodland Hills, Calif. Dr Freund is a clinical professor of ophthalmology at NYU School of Medicine and practices at Vitreous Retina Macula Consultants of New York.
1. Gass JD. Acute Posterior Multifocal Placoid Pigment Epitheliopathy. Arch Ophthalmol 1968;80:177-85.
2. Goldenberg D, Habot-Wilner Z, Loewenstein A, Goldstein M. Spectral domain optical coherence tomography classification of acute posterior multifocal placoid pigment epitheliopathy. Retina 2012;32:1403-10.
3. Hirano Y, Yasukawa T, Nagai H, Ogura Y. Spatio-temporal understanding of the pathology of acute posterior multifocal placoid pigment epitheliopathy. Jpn J Ophthalmol 2012;56(4):371-4.
4. Scheufele TA, Witkin AJ, Schocket LS, Rogers AH, et al. Photoreceptor atrophy in acute posterior multifocal placoid pigment epitheliopathy demonstrated by optical coherence tomography. Retina 2005;25:1109-12.
5. Cheung CM, Yeo IY, Koh A. Photoreceptor changes in acute and resolved acute posterior multifocal placoid pigment epitheliopathy documented by spectral-domain optical coherence tomography. Arch Ophthalmol 2010;128:644-6.
6. Souka AA, Hillenkamp J, Gora F, Gabel VP, Framme C. Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefe’s Arch Clin Exp Ophthalmol 2006;244:1219-23.
7. O’Halloran HS, Berger JR, Lee WB, Robertson DM, et al. Acute multifocal placoid pigment epitheliopathy and central nervous system involvement: nine new cases and a review of the literature. Ophthalmology 2001;108:861-8.
8. Gass JDM. Stereoscopic atlas of macular diseases: diagnosis and treatment. 3rd ed. vol 1. St Louis: Mosby, 1987.
9. Lim WK, Buggage RR, Nussenblatt RB. Serpiginous Choroiditis. Surv Ophthalmol 2005;50(3):231-44.
10. Hardy RA, Schatz H. Macular geographic helicoid choroidopathy. Arch Ophthalmol 1987;105:1237-42.
11. Hamilton AM, Bird AC. Geographical choroidopathy. Br J Ophthalmol 1974;58:784-97.
12. Christmas NJ, Oh KT, Oh DM, Folk JC. Long-term follow-up of patients with serpinginous choroiditis. Retina 2002;22:550-6.
13. van Velthoven ME, Ongkosuwito JV, Verbraak FD, Schlingemann RO, de Smet MD. Combined en-face optical coherence tomography and confocal ophthalmoscopy findings in active multifocal and serpiginous chorioretinitis. Am J Ophthalmol 2006;141:972-5.
14. Bansal R, Gupta A, Gupta V. Imaging in the diagnosis and management of serpiginous choroiditis. Int Ophthalmol Clin 2012;52(4):229-36.
15. Carreño E, Portero A, Herreras JM, López MI. Assesment of fundus autofluorescence in serpiginous and serpiginous-like choroidopathy. Eye (Lond) 2012;26:1232-6.
16. Yeh S, Forooghian F, Wong WT, Faia LJ, et al. Fundus autofluorescence imaging of the white dot syndromes. Arch Ophthalmol 2010;128:46-56.
17. Blumenkranz MS, Gass JD, Clarkson JG. Atypical serpiginous choroiditis. Arch Ophthalmol 1982;100:1773-5.
18. Lee DK, Suhler EB, Augustin W, Buggage RR. Serpiginous choroidopathy presenting as choroidal neovascularisation. Br J Ophthalmol 2003;87:1184-5.
19. Jampol LM, Orth D, Daily MJ, Rabb MF. Subretinal neovascularization with geographic (serpiginous) choroiditis. Am J Ophthalmol 1979;88:683-9.
20. Gupta V, Agarwal A, Gupta A, Bambery P, Narang S. Clinical characteristics of serpiginous choroidopathy in North India. Am J Ophthalmol 2002;134:47-56.
21. Song MH, Roh YJ. Intravitreal ranibizumab for choroidal neovascularisation in serpiginous choroiditis. Eye (Lond) 2009;23:1873-5.
22. Mansour AM, Jampol LM, Packo KH, Hrisomalos NF. Macular serpiginous choroiditis. Retina 1988;8:125-31.
23. Sahu DK, Rawoof A, Sujatha B. Macular serpiginous choroiditis. Indian J Ophthalmol 2002;50(3):189-96.
24. Bansal R, Gupta A, Gupta V, Dogra MR, Sharma A, Bambery P. Tubercular serpiginous-like choroiditis presenting as multifocal serpiginoid choroiditis. Ophthalmology 2012;119:2334-42.
25. Gupta V, Bansal R, Gupta A. Continuous progression of tubercular serpiginous-like choroiditis after initiating antituberculosis treatment. Am J Ophthalmol 2011;152(5):857-63.
26. Gupta V, Gupta A, Arora S, Bambery P, Dogra MR, Agarwal A. Presumed tubercular serpiginouslike choroiditis: Clinical presentations and management. Ophthalmology 2003;110:1744-9.
27. Jones BE, Jampol LM, Yannuzzi LA, Tittl M, et al. Relentless placoid chorioretinitis: A new entity or an unusual variant of serpiginous chorioretinitis? Arch Ophthalmol 2000;118:931-8.
28. Jyotirmay B, Jafferji SS, Sudharshan S, Kalpana B. Clinical profile, treatment, and visual outcome of ampiginous choroiditis. Ocul Immunol Inflamm 2010;18:46-51.
29. Biswas J, Narain S, Das D, Ganesh SK. Pattern of uveitis in a referral uveitis clinic in India. Int Ophthalmol 1996-1997;20(4):223-8.
30. Bouchenaki N, Cimino L, Auer C, Tao Tran V, Herbort CP. Assessment and classification of choroidal vasculitis in posterior uveitis using indocyanine green angiography. Klin Monbl Augenheilkd 2002;219:243-9.
31. Amer R, Florescu T. Optical coherence tomography in relentless placoid chorioretinitis. Clin Experiment Ophthalmol 2008;36(4):388-90.
32. Golchet PR, Jampol LM, Wilson D, Yannuzzi LA, Ober M, Stroh E. Persistent placoid maculopathy: A new clinical entity. Trans Am Ophthalmol Soc 2006;104:108-20.
33. Golchet PR, Jampol LM, Wilson D, Yannuzzi LA, Ober M, Stroh E. Persistent placoid maculopathy: A new clinical entity. Ophthalmology 2007;114:1530-40.
34. Kovach JL. Persistent placoid maculopathy imaged with spectral domain OCT and autofluorescence. Ophthalmic Surg Lasers Imaging 2010 Nov-Dec;41 Suppl:S101-3
35. Parodi MB, Iacono P, Bandello F. Juxtafoveal choroidal neovascularization secondary to persistent placoid maculopathy treated with intravitreal bevacizumab. Ocul Immunol Inflamm 2010;18(5):399-401.
36. Pearlman RB, Golchet PR, Feldmann MG, Yannuzzi LA, et al. Increased prevalence of autoimmunity in patients with white spot syndromes and their family members. Arch Ophthalmol 2009;127:869-74.