Traditional models of treating glaucoma have centered on the one factor we know reduces the risk of progression for most patients: lowering intraocular pressure. However, with the advent of gene therapy as a potentially viable way to preserve vision, new modalities for managing glaucoma may soon become available.
As a researcher and clinician, I’m very interested in finding ways to keep retinal ganglion cells and neurons alive, and gene therapy is showing promise as a way to do that. However, many clinicians aren’t well-acquainted with this technology, for two reasons. First, it’s not widely used in practice right now because of the limited patient populations eligible for the few gene therapy treatments that have been approved by the U.S. Food and Drug Administration. Second, many clinicians haven’t studied this topic since medical school.
With that in mind, I’d like to provide a brief review of the concepts behind gene therapy; discuss the use of gene therapy in current human trials; and review a neuroprotective therapy currently in development that’s relevant to glaucoma management. The latter, which we’re working on in our lab, is in the pre-clinical development phase but is quickly moving into the clinical phase.
 |
Gene Therapy: The Basics
Every cell in our body contains genes made up of DNA, the genetic material that humans and almost every other organism uses to multiply themselves, as well as to multiply the cells that make up their bodies. Some of the DNA encodes information that allows the genes to construct proteins; those proteins help to build the body and help it function. The catch is that mutations in our genes can develop over time or be inherited. Those mutations may then create abnormal proteins that can impact our bodies and our health.
A classic example is one most of us learned about in medical school: sickle cell anemia. In this disease, an inherited mutation causes a subtle change in the way a key protein in red blood cells is folded. That, in turn, alters the way the cells are shaped, causing them to act differently, resulting in the disease.
Gene therapy involves introducing, removing or changing genetic material within cells to repair or compensate for the loss of function in a gene. Altering genetic material allows us to increase or overexpress proteins that will fight a disease, or even produce new proteins for this purpose. We’ve been doing this in the laboratory for a while now by using what’s called “small interfering RNA,” or siRNA, to interfere with the expression of specific genes, preventing them from forming proteins.
This is the science that allowed the creation of the COVID-19 vaccines. These vaccines included genetic material that taught our cells to make a protein resembling the spikes on the COVID virus. That protein was sufficient to get our immune systems to generate a defensive response that allowed us to avoid getting tremendously sick from the coronavirus.
As you can see, the idea of producing new proteins or modified proteins to fight disease is already at the cutting edge of vaccine therapy—and we’re getting closer to using similar technology to treat and potentially cure eye diseases.
 |
 |
Approaches to Gene Therapy
Gene therapy can be used to treat human disease via:
• Gene replacement. Jean Bennett, MD, PhD, is a pioneer in this approach. She created Luxturna, the first licensed gene therapy for Leber’s Congenital Amaurosis (LCA), a rare, inherited eye disorder. (I had the good fortune to be mentored by Dr. Bennett here at the University of Pennsylvania.) This approach replaces a mutated gene that’s not working with a working gene.
• Gene silencing. This approach delivers messenger RNA that stops the production of a protein.
• Gene editing. This uses the so-called CRISPR technology, which has been written about a lot lately. (CRISPR stands for “clustered regularly interspaced short palindromic repeats.” It uses repeated letters of the genetic code to tell an enzyme exactly where to cut a strand of DNA.) The CRISPR technique uses a guide RNA to potentially alter a gene within the patient’s genome. It’s a very effective technique, and we’ll likely see more of it used in relation to treating human disease.
• Gene addition. This technique causes overexpression of a gene that can positively impact a disease state. This technique is useful when a protein is already being made, but we need more of it so that cells can survive or prevent the disease altogether. We’re working on some treatments involving this approach here at the University of Pennsylvania.
Getting Material into the Cells
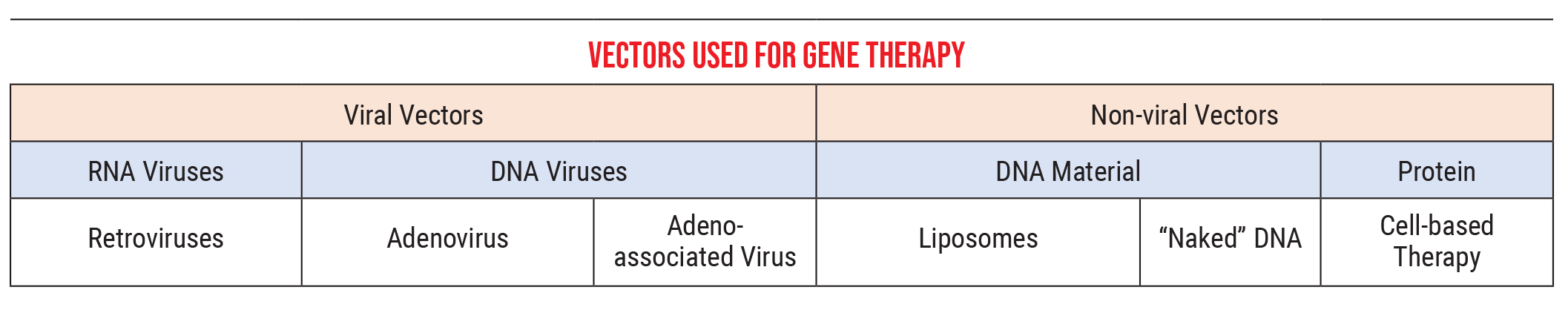
The other part of this that’s important to understand is the methods we use to introduce this genetic material into the cells. Genetic material is delivered by what we call vectors, courier particles that carry the new genetic information to the cells.
Currently, we primarily use two kinds of vectors:
• Viral vectors. These can be RNA viruses, such as retroviruses, or DNA viruses, which would include adenoviruses and adeno-associated viruses. These viruses introduce our genetic material into a cell in much the same way a standard virus would. (In our lab we’re using mostly adeno-associated viruses, because adenoviruses cause some immune-system-related problems not seen with adeno-associated viruses. We deliver the adeno-associated virus material via an intraocular injection.)
• Nonviral vectors. These can be DNA-related material such as liposomes, “naked” DNA or simple proteins, an approach that’s also referred to as “cell-based therapy.” The genetic material that we got from the COVID vaccines was delivered in liposome form, for example.
As a point of clarification, retroviruses (one of the viral vector options) are a type of RNA virus that can take RNA and turn it into DNA. For example, HIV is a retrovirus that can do this. This ability contradicts one of the central dogmas many of us learned in biology class, that DNA is stable and can generate RNA, which then creates proteins, while RNA generally can’t be turned back into DNA. In actuality, some retroviruses can transform RNA back into DNA, and some can even insert that DNA into your genome. (We don’t use those in our lab, because it’s hard to control where the genes end up in your genome. We wouldn’t want this to cause a loss of function in some area unrelated to the disease we’re trying to treat. It’s more straightforward to deliver stable DNA using an adeno-associated virus or adenovirus.)
Once you get the material to the cells, there are several ways in which the vector can interact with the genetic material. The choice you make in this area helps to answer other important questions. For example, once the material gets into the cell, do you want it to be turned on in every cell in your body (referred to as “ubiquitous” delivery), or only in certain specific cells (“cell-specific” delivery)?
This is managed by what’s called the promoter sequence, a sequence of genetic information that sits in front of the therapeutic gene. It’s like a very specific key to a door that enables the process to be turned on or off, regulating whether the cell starts generating mRNA and protein. If the key doesn’t fit, the door won’t open. The promoter can be a master key, starting the process in every cell, or a specific key that only starts it in one particular type of cell. This gives us a way to control which cells end up producing the protein.
Of course, additional cell selectivity can be achieved by proximity—where you inject the gene therapy. (This is a key part of what makes Luxturna an effective therapy.) This is an important factor when injecting things into the eye, because the eye is a tremendously immuno-privileged organ; there’s very little crossover into the body.
 |
Our Work Protecting Cells
As noted earlier, our lab is working on genetic treatments that may help preserve vision in glaucoma patients, using adeno-associated viruses to deliver genetic material to retinal ganglion cells. We’re using this method to get the cells to produce a protein that’s been shown to be protective of ganglion cells: SIRT1.
The preclinical studies of our treatment were done using an animal model. One of our early experiments, intended to demonstrate the validity of this approach, involved delivering genetic material designed to make cells in the retina produce a fluorescent green protein not normally produced by these cells. To introduce the material into a rodent eye, we’d cut down the conjunctiva and insert our vector material into a small syringe; then we’d insert the needle just under the lens, being careful not to hit it. At first, we were able to get about 42 percent of the cells to produce the protein using this approach. Since then, we’ve been able to increase the percentage of cells producing the desired protein significantly.
In any case, part of the importance of this experiment is that the cells expressing the protein can be counted. That’s crucial, because that allows us to quantify the strength of the vector, and to test it for safety; we can see if the vector is reaching the cells we want it to reach. In this case, we wanted the vector to reach cells in the retinal ganglion cell layer, and histological studies have demonstrated that very few cells outside the RGC layer ended up producing the fluorescent green protein.
Of course, the green fluorescent protein won’t cure any human disease, but there are proteins that we can use to influence human disease, such as SIRT1. My senior mentor and main collaborator, Kenneth S. Shindler, MD, PhD, has conducted numerous studies demonstrating SIRT1’s ability to keep RGCs alive in the face of injury from optic neuritis and optic nerve crush.1-7 So we thought: What if we could make these cells create more SIRT1 using gene therapy in a cell-specific manner? Our hope was that the retinal ganglion cells expressing SIRT1 would be able to avert some of the damage caused by injuries to the optic nerve.
Our studies demonstrated that the treatment did cause the ganglion cells to increase their production of SIRT1, as much as a thousandfold. So the next question was, would this be protective if the intraocular pressure was raised?
To find out, we injected microbeads into the anterior chamber of the eye; the beads then ended up in the iridocorneal angle, blocking fluid access to the trabecular meshwork. This caused the pressure to rise, and as their IOP was sustained and elevated, the mice began to lose vision, just as people do. (Importantly, that loss of vision correlates very well with the loss of retinal ganglion cells, giving us a structure/function correlation.) We also had a control group of mice that received BSS instead of microbeads. Then, some mice received our therapeutic injection, while others received a sham injection consisting of the genetic material than causes the production of green fluorescent proteins—which has no therapeutic capacity—instead of SIRT1.
The therapy was indeed successful, as shown in the graphs. The mice with elevated pressure that didn’t receive therapy lost vision over time, but those receiving the genetic treatment had prolonged visual capacity. Counting the surviving retinal ganglion cells also showed the same result; more RGCs survived when the eye received the genetic therapy. That suggests two things: first, that RGCs are responsible for maintaining vision in the eye, and second, that our genetic SIRT1 therapy can be protective against IOP-related cell damage.
Further confirmation was provided by measuring axon density in the eyes. The axons are the fibers that take the information from the retinal ganglion cells to the brain. Normally, the axons are numerous, dense and healthy. The mice that received microbeads without our therapy showed a loss of axon density, while those receiving the therapy showed a reduced level of axon damage. (These data have been submitted for publication.)
Just Getting Started
For a while now, gene therapy has represented a true paradigm shift in the treatment of optic neuropathies. So far, the impact has been limited; the successful treatment of Leber’s, for example, only affects a small number of patients. (The disease is terrible, but it’s not a common optic neuropathy.) However, treatments addressing more common optic neuropathies like glaucoma, ischemic optic neuropathy and inflammatory optic neuropathies are in the works. Our lab has shown that using genetic modification to enhance SIRT1 production can be very effective at preventing ganglion cell loss and degeneration in multiple models of optic nerve disease, and many other efforts along similar lines are currently under way.
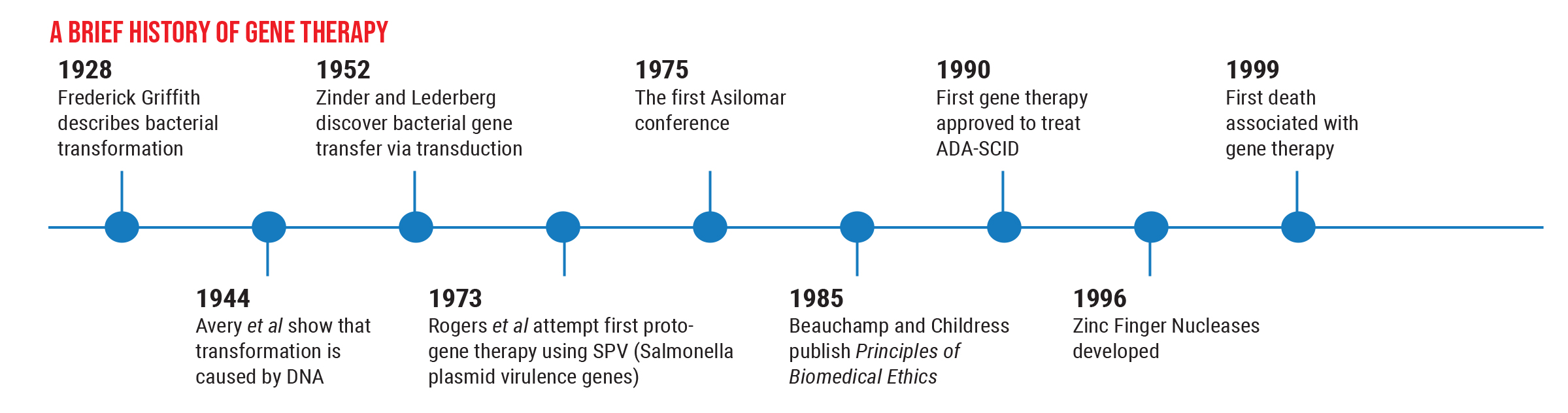
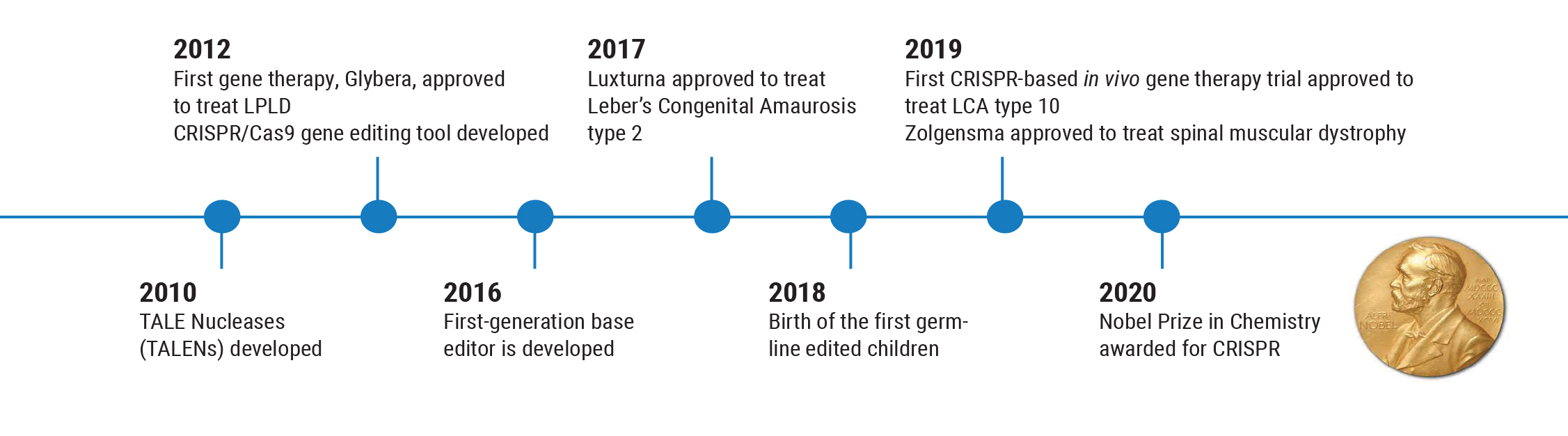
However, these options are still in the preclinical stage, and we want to proceed with caution. The history of this field illustrates why this is so important. Gene therapy has been successfully used to treat human disease back as far as 1990, but nine years later, the first death connected with gene therapy occurred. This caused everyone to backpedal. As a result of that death, many regulations were put in place to ensure a greater level of safety for patients.
Partly for that reason, the next gene therapy—Glybera (alipogene tiparvovec), designed to reverse lipoprotein lipase deficiency that can cause severe pancreatitis—wasn’t approved until 2012. This was followed by another five-year gap before Luxturna was approved to treat LCA Type 2 retinal degeneration in 2017. Those circumstances have led us to create more careful, thorough trials before we use a gene therapy in humans, to make sure it’s both safe and effective.
This brings us to a very practical clinical issue. Many clinicians have worked with patients who’ve had the terrible experience of losing vision because of optic neuropathies or glaucoma. Today, as awareness of gene therapy has grown, patients in this situation almost always ask if there’s a clinical trial they can get into. Unfortunately, there are individuals out there conducting “clinical trials” that aren’t approved or monitored, giving hope to these patients while putting them at risk.
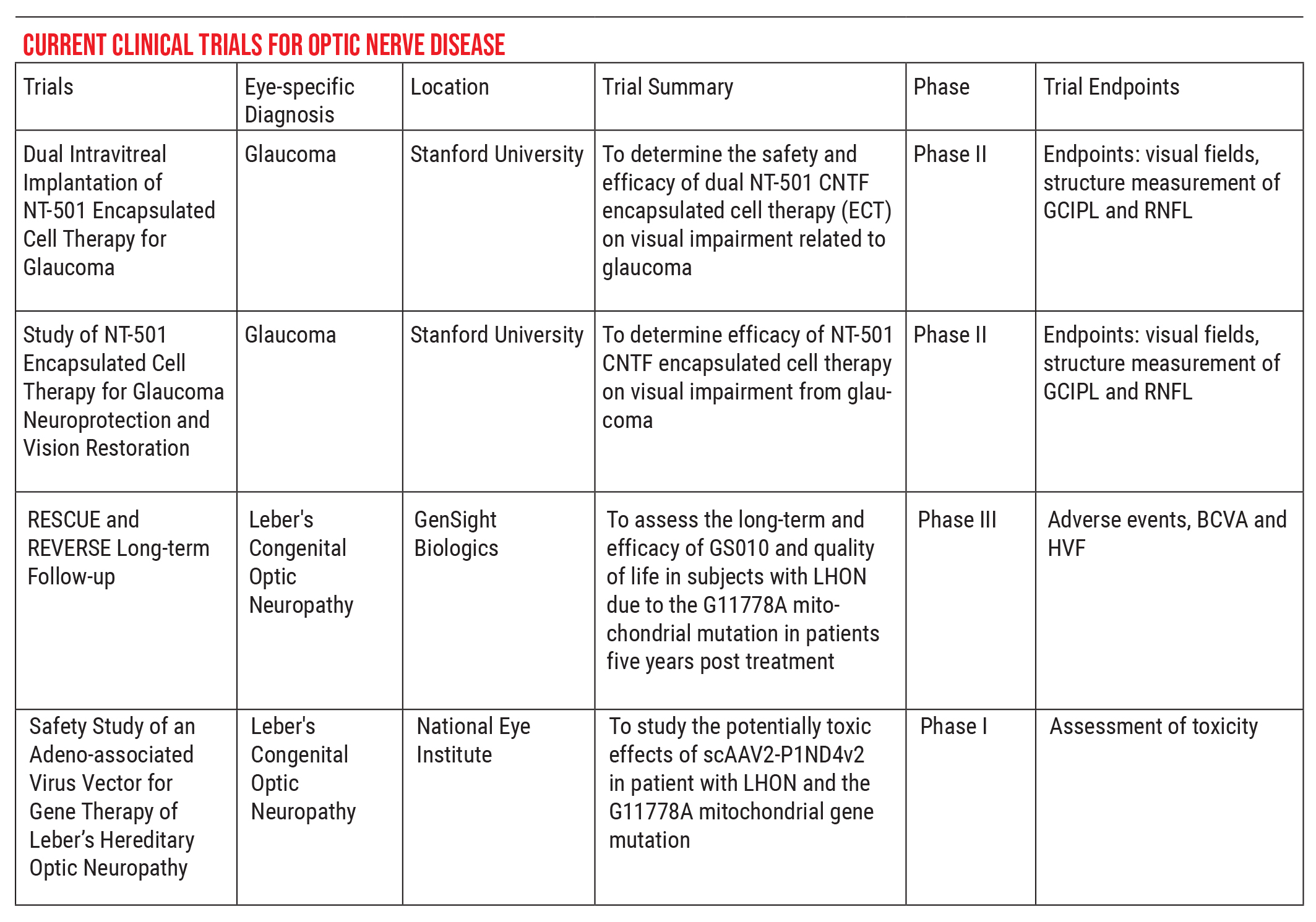
For that reason, part of our job is to help steer these patients in the right direction. If you’re looking for a trial that has regulatory oversight, I recommend visiting asgct.org/clinicaltrials, a database showing currently active clinical trials. For example, there are currently two clinical trials relating to glaucoma, both out of Stanford, and two for Leber’s hereditary optic neuropathy, the RESCUE and REVERSE trials. The people in those trials are very stringently selected, to ensure that no harm can come to them, and the data can be used to monitor meaningful effects.
So, when patients come to me asking about studies or investigators they’ve heard about in which healthy eyes are being injected with genes or cell-based therapies, I go to this website to make sure the study has been submitted. If it hasn’t, then I have to have another kind of conversation with the patient. I have to explain that some trials out there are not necessarily safe, and not in the best interests of the patient. Some patients may be adamant about proceeding, but at least I can warn them about the risk they may be taking.
We’re on the verge of a new era in treatment for glaucoma and other optic neuropathies. These emerging genetic technologies for treatment are going to move us beyond just prescribing a drop to lower the pressure. Within a few years our focus may shift to strategies that help to keep those vision-providing retinal cells happy and alive. That’s a revolution we can all look forward to.
This article has no commercial sponsorship.
Dr. Singh is a professor of ophthalmology and chief of the Glaucoma Division at Stanford University School of Medicine. Dr. Netland is Vernah Scott Moyston Professor and Chair at the University of Virginia in Charlottesville.
Dr. Ross is an assistant professor of ophthalmology and neurology at the University of Pennsylvania and Scheie Eye Institute, in the divisions of glaucoma and neuro-ophthalmology. She’s received research funding from the National Institutes for Health and Gyroscope Therapeutics, recently acquired by Novartis.
1. Ross AG, McDougald DS, Khan RS, et al. Rescue of retinal ganglion cells in optic nerve injury using cell-selective AAV mediated delivery of SIRT1. Gene Ther 2021;28:5:256-264.
2. McDougald DS, Dine KE, Zezulin AU, Bennett J, Shindler KS. SIRT1 and NRF2 gene transfer mediate distinct neuroprotective effects upon retinal ganglion cell survival and function in experimental optic neuritis. Invest Ophthalmol Vis Sci 2018;59:3:1212-1220.
3. Zuo L, Khan RS, Lee V, et al. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Invest Ophthalmol Vis Sci 2013;54:7:5097-102.
4. Khan RS, Fonseca-Kelly Z, Callinan C, et al. SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front Cell Neurosci 2012;6:63.
5. Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest Ophthalmol Vis Sci 2007;48:8:3602-9.
6. Khan RS, Dine K, Das Sarma J, Shindler KS. SIRT1 activating compounds reduce oxidative stress mediated neuronal loss in viral induced CNS demyelinating disease. Acta Neuropathol Commun 2014;2:3.
7. Ross AG, Chaqour B, McDougald DS, et al. Selective upregulation of SIRT1 expression in retinal ganglion cells by aav-mediated gene delivery increases neuronal cell survival and alleviates axon demyelination associated with optic neuritis. Biomolecules 2022;12:6:830.


