Given the ocular examination, the working diagnosis was Peters anomaly. The differential diagnosis of a congenitally cloudy cornea also includes congenital hereditary endothelial dystrophy (CHED), Axenfeld-Rieger spectrum, keratitis (e.g., herpes simplex), metabolic disorders, forceps trauma and congenital glaucoma, as well as alternative anterior segment dysgeneses.
The decision was made to pursue an examination under anesthesia which confirmed these findings and was again consistent with Peters anomaly in both eyes. Ultrasound biomicroscopy (See Figures 1 and 2 on the previous page) demonstrated the iridocorneal adhesions in both eyes, as well as a central posterior corneal scalloped defect in the left eye. There was no evidence of glaucoma in either eye.
The abnormalities in the left eye were felt to be visually significant. Therefore, due to the potential for amblyopia, the patient underwent penetrating keratoplasty at four months of age. The patient was left phakic. Follow-up visits focused on glaucoma screening in both eyes and visual rehabilitation for amblyopia in the left eye. Fortunately, examination under anesthesia at age 1 did not reveal evidence of glaucoma in either eye, and the corneal graft in the left eye was clear (See Figure 3). At one year the vision in the left eye was central, steady and non-maintained.
Discussion
Peters’ anomaly results from a sequence of events that unfold from a failure of the lens placode to separate from the overlying primordial cornea during the fourth to seventh week of embryogenesis.1 Consequently, there is a phenotypically heterogeneous spectrum of disease which presents with corneal opacification and is frequently associated with iridocorneal and/or corneolenticular adhesions, glaucoma, sclerocornea or microphthalmia.
Peters’ anomaly is an anterior segment dysgenesis associated with mutations in key genes that are responsible for anterior segment embryogenesis, such as PAX6, PITX2, FOXC1 and CYP1B1. Recent studies have also shown associations between isolated Peters’ anomaly and mutations in HCCS, NDP and SLC4A11.1 However, there are certainly other genes yet to be identified. Correlations between specific mutations and phenotypic features have not proven to be straightforward, suggesting a complex interaction between other genetic and epigenetic factors.2
Historically, Peters’ anomaly has been split into two categories: type I and type II. Type I is characterized by iridocorneal adhesions, while type II exhibits corneolenticular adhesions and cataract. However, these classifications have been regarded as inadequate as they do not describe the clinical severity of the disease, which is often attributable to the extent of associated corneal opacity.3 Various studies have attempted to provide a systematic approach for gauging disease severity. One study suggested that severe disease is characterized by a corneal opacity extending over half of the cornea, cataracts, corneolenticular adhesions, persistent hyperplastic primary vitreous or microphthalmia.3 Ultimately, the primary factor driving treatment is the degree of potential visual compromise.
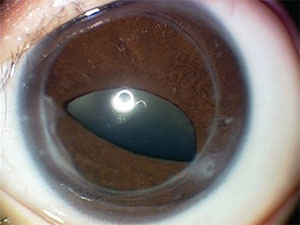 |
| Figure 3. Left eye nine months after penetrating keratoplasty, showing a clear corneal graft and iridocorneal adhesions nasally and temporally to the graft-host junction. |
Timing and modality of treatment is a debated topic. The impetus for early intervention is the prevention of deprivation amblyopia while, at the same time, waiting until the child is an appropriate surgical candidate. Medical management can be considered for cases where the extent of the corneal opacity would not be expected to cause significant visual impairment.4 The use of mydriatics has been proposed to allow visual input to pass around the opacity to prevent amblyopia.3,4 A limited, side-by-side comparison of medical versus surgical intervention found that patients grouped for medical management had superior visual outcomes. However, the study’s authors noted that this finding was likely confounded by the cohort of medically managed patients having less-severe disease at the time of presentation.
Surgical intervention typically includes a penetrating keratoplasty with or without concurrent cataract extraction and adhesion lysis. Reports of penetrating keratoplasty outcomes in the pediatric population vary greatly in current literature, with graft success rates ranging from 18 to 90 percent.5,6 One group of researchers suggests that this broad range of postoperative outcomes likely relates to disease severity, given the higher graft success rates with type I as opposed to type II Peters’ anomaly. A recent study reviewing graft survival rates of primary penetrating keratoplasty for pediatric patients demonstrated a mean survival rate of 90 percent in eyes without glaucoma. Only 52 percent of grafts survived at one year in eyes that had developed concomitant glaucoma.6 This study implicated the presence of glaucoma and a history of concurrent operations at the time of primary penetrating keratoplasty as the largest factors contributing to graft failure.6
Visual outcomes across studies also vary considerably. A paper from 2009 reviewed long-term visual outcomes for pediatric patients who underwent one or more keratoplasties. The researchers observed that 29 percent of eyes achieved a long-term visual acuity better than 20/400, while 38 percent of patients had outcomes with LP or NLP vision.7 Further, the authors found that early surgical intervention provided little to no benefit for long-term visual outcomes.7 It’s important to note that many of these studies are limited by the heterogeneous nature of Peters’ anomaly and variable timing of surgery, making patient comparison difficult.
Perhaps the most clinically challenging complication of Peters’ anomaly is glaucoma. It has been estimated that 50 to 70 percent of patients with anterior segment dysgenesis will develop glaucoma.2,8 Research has demonstrated that after a review of 126 glaucoma procedures on 34 eyes with findings of Peters’ anomaly, only 32 percent of them had satisfactory intraocular pressure control at the 11-year follow-up, and over a third of those patients required multiple procedures. Overall outcomes were poor, with roughly half of the operated eyes having only light perception vision or being lost to phthisis or retinal detachment.9
Ultimately, patients presenting with findings of Peters’ anomaly require a multidisciplinary approach to evaluation and treatment. This includes routine glaucoma screening for any child presenting with Peters’ anomaly, regardless of severity, with or without a history of penetrating keratoplasty. Additionally, associated systemic manifestations such as skeletal dysplasia may be present. This combination of ocular and systemic findings is referred to as Peters’ Plus syndrome, and it’s important for these patients to have a thorough examination by their pediatrician. Genetic counseling is also an important component in caring for patients with Peters’ anomaly, especially with the availability of molecular genetic testing. Therefore, management of this complex disease demands coordinated care shared among pediatricians, pediatric ophthalmologists, cornea specialists and genetic counselors, as well as glaucoma specialists. REVIEW
1. Weh E, Reis L, Happ H, Levin A, Wheeler P, David K, Carney E, Angle B, Hauser N, Semina E. Whole exome sequence analysis of Peters’ anomaly. Hum Genet 2014;133:1497-1511.
2. Ito Y, Walter M. Genomics and anterior segment dysgenesis: A review. Clinical and Experimental Ophthalmology 2014;42:13–24.
3. Chang JW, Kim JH, Kim SJ, et al. Long-term clinical course and visual outcome associated with Peters’’ anomaly. Eye (Lond) 2012;26:9:1237-42.
4. Bhandari R, Ferri S, Whittaker B, et al. Peters’ Anomaly: Review of Literature. Cornea 2011;30:8:939-944.
5. Kim YW, Choi HJ, Kim MK, et al. Clinical outcome of penetrating keratoplasty in patients 5 years or younger: Peters’ anomaly versus sclerocornea. Cornea 2013;32:11:1432-6.
6. Karadag R, Chan TC, Azari AA, Nagra PK, Hammersmith KM, Rapuano CJ. Survival of Primary Penetrating Keratoplasty in Children Am J Ophthalmol 2016;171;16:95-100.
7. Abdolrahimzadeh, S, Fameli V, Mollo R, et al. Rare diseases leading to childhood glaucoma: Epidemiology, pathophysiogenesis, and management. Biomed Res Int 2015;2015:781294.
8. Yang LL, Lambert SR, Drews-Botsch C, et al. Long-term visual outcome of penetrating keratoplasty in infants and children with Peters’ anomaly. J AAPOS 2009;13:2:175-80.
9. Yang LL, Lambert SR, Lynn MJ, et al. Surgical management of glaucoma in infants and children with Peters’’ anomaly: Long-term structural and functional outcome. Ophthalmology 2004;111:1:112-7.


