Rubor, tumor, calor, dolor (redness, swelling, heat and pain): The Roman encyclopedist and physician Aulus Cornelius Celsus first documented these classic signs of acute inflammation in the first century. Then, in 1870, Rudolf Virchow, "the father of pathology," added functio laesa, or disturbance of function.1 Although these descriptors remain accurate, recent research into inflammation at the cellular and molecular levels has revealed the complexity of this process.
Acute inflammation is a normal systemic response to injury or infection. The stimulus initiates a signaling cascade designed to kill invading microorganisms, protect tissue from damage and heal damaged tissue. The signaling originates in cells already present in the tissue (epithelial cells, macrophages, endothelial cells, dendritic cells, mast cells) through the release of histamine, cytokines, chemokines and eicosanoids. A fundamental aspect of the inflammatory response is the nature of the communication among the different specialized cells. Rather than a simple linear process, the tissue response to injury is orchestrated and highly complex, involving a large signaling network among diverse cell types.2 This degree of complexity has given rise to systems biology, an analytical modeling approach that combines elements of mathematics, engineering and biology.3 Acute inflammation, a highly regulated response, mediated by short-lived signals, was one of the first areas where systems biology demonstrated predictive value.4 Models of sepsis provided insight into whether the anti-inflammatory (healing) phase was a subsequent and compensatory response to the initial pro-inflammatory phase, or whether the two responses were initiated concurrently; the modeling supported the latter.4 Considerable evidence now supports the idea that the return to normal cellular activity following acute inflammation is active rather than passive, i.e., a coordinated program of resolution is initiated in the first few hours after an inflammatory response begins.5 When something goes wrong and the inflammation becomes chronic, the body's continual response can produce simultaneous tissue destruction and attempts at repair that can lead to hyperplasia, angiogenesis, neovascularization, fibrosis, ulceration and even cancer.
The Wound That Doesn't Heal
In chronic inflammation, the initiating stimulus persists or the mechanism required for reversing the pro-inflammatory signaling fails. Neutrophils infiltrating sites of injury, in response to pro-inflammatory cytokines and chemokines, mediate acute inflammation. In contrast, chronic inflammation involves lymphocytes, macrophages, neutrophils and the production of a complex mélange of growth factors and cytokines that cause general hyperplasia, fibrosis, angiogenesis and neovascularization.
Before discussing chronic inflammation in age-related macular degeneration, it may be informative to consider chronic inflammation in other tissues in order to draw some parallels. Exposure of healthy tissue to a high dose of ionizing radiation can cause delayed side effects that have been linked to a chronic inflammatory process. Normal tissue side effects, which may occur long after radiation exposure, are the limiting factor in the dose of radiation that can be delivered to a tumor. "A perpetual cascade of cytokines" throughout the six-month latent period following radiation exposure and prior to the development of side effects was initially demonstrated in the mouse lung,6 and shortly thereafter in the mouse brain7 and the rat small intestine.8
A signaling cascade occurring after the initial insult to the tissue represents an opportunity for pharmacologic intervention. Indeed, a statin was effective in reducing late-appearing side effects in irradiated mouse lung.9 Statins are also potent anti-inflammatory agents, able to reduce chemokine expression and inflammatory cell recruitment.10 Lovastatin (10 mg/kg, three times per week beginning eight weeks post-irradiation) improved the survival rate, decreased the collagen content and reduced the degree of macrophage and lymphocyte infiltration in the lungs of mice at 12, 16 and 24 weeks after irradiation.9 These studies were among the first to demonstrate a causative link between chronic inflammation and late-appearing tissue damage. The current understanding is that late-appearing normal tissue damage is not necessarily the inevitable result of the initial acute radiation damage to a critical cell population, but rather the consequence of the body's subsequent response. Radiation-induced effects in normal tissues have been described as a "wound that does not heal."11
But what provides the chronic stimulus that drives the chronic inflammation? The gradual loss of a damaged population of long-lived cells has long been assumed to play a role, though whether the critical cell population is the vascular endothelium or functional parenchymal cells continues to be debated.12
Strong evidence for the former comes from a study in the central nervous system. A progressive decrease in vascular endothelial cell density was demonstrated in irradiated rat brain during the one-year latent period before the development of white matter necrosis.13 Selective protection of the vascular endothelium by having the radical scavenger amifostine present only in the blood at the time of irradiation reduced both the extent of endothelial cell loss and the incidence of white matter necrosis. Vascular endothelial cells have turnover times measured in months.14 As individual cells proceed through the cell cycle, if DNA damage from prior radiation exposure is too extensive, DNA replication or mitosis fails and the cells undergo an apoptotic death.15 Signals released from the apoptotic endothelial cells, or from neighboring endothelial cells in response to an apoptotic neighbor, are postulated to be the chronic inflammatory stimulus.
|
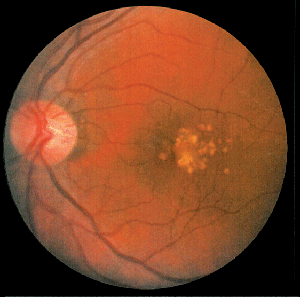
What About the Eye?
AMD is a complex, progressive disorder of the central retina characterized by the formation of sub-retinal deposits (drusen), damage to and loss of retinal pigment epithelial cells and photoreceptors, and, eventually, choroidal neovascularization. Though the mechanism of the progression process in AMD remains unclear, recent progress in genetic analyses and studies in gene-knockout animal models provide insights that indicate chronic inflammation may be one of the fundamental causes of the overall pathology.
The retina is a complex tissue with high metabolic activity, high blood flow, areas of high oxygen tension and high levels of light-absorbing chromophores. In the normal retina, RPE cells phagocytose photoreceptor outer segments as they age and accumulate oxidative damage byproducts. The bleached chromophore all-trans-retinal is normally cleared from photoreceptor discs and regenerated in a process referred to as the visual cycle. A small fraction of the all-trans-retinal reacts with endogenous amines to create fluorescent chromophores that progressively accumulate in or below the RPE cells as lipofuscin deposits. Some of the all-trans-retinal condensation products, such as the bis-retinoid compound A2E, react with light and oxygen creating chemically reactive intermediates, which can be locally toxic; "oxidative stress" has long been discussed in relation to the development of AMD.
Drusen are deposits accumulating between the basal surface of the RPE and Bruch's membrane. Drusen are a hallmark of early, or dry, AMD and a significant risk factor for progression to wet AMD. Drusen include remnants of RPE cells and a variety of immune system-related molecules. In 2001 and 2002, researchers first suggested that the immune system recognizes drusen as extracellular deposits of foreign material, and initiates an inflammatory response through the alternative complement pathway.16,17 Currently, considerable evidence supports this hypothesis.18-22 The complement system is a tightly controlled process responsible for killing invading microorganisms. Genetic linkage studies in AMD patients identified defects in several complement proteins responsible for regulation of the complement activation process, which dramatically increased the likelihood of developing AMD.19,20,22 Researchers have proposed that removal of oxidatively damaged intracellular proteins—including mitochondrial proteins—from aging RPE cells through an exocytic process may be involved in the formation of drusen.23 Normally, the complement inhibitor, complement factor H (CFH), protects released exosomes from complement attack until they can be cleared. In patients with a genetic defect in the CFH gene, a known risk factor for AMD, disrupted exosomes may release intracellular proteins into the extracellular environment. Increased exosome markers were found surrounding Bruch's membrane in the aged mouse eye, and in drusen from AMD patients.23
Although advanced age is a risk factor for AMD, Stargardt's disease, a hereditary condition characterized by juvenile onset, rapid progression and a poor visual outcome, shares some of the characteristic features of AMD, including accumulation of lipofuscin in RPE cells and degeneration of photoreceptors. Choroidal neovascularization occurs in only a small subset of cases. The defect associated with Stargardt's disease is in the gene that encodes a retina-specific transport protein in the rims of rod and cone outer segment discs (ATP binding cassette transporter A4, ABCA4). ABCA4 transfers all-trans-retinal from the inside to the outside of disc membranes.24 This is an important transporter with a main function to facilitate the visual cycle by speeding up the removal of all-trans-retinal from photoreceptors. In a murine ABCA4 knockout model, mice developed elevated levels of all-trans-retinal, lipofuscin and A2E.24 Subsequent studies have shown that light exposure leads to the formation of A2E epoxides and oxiranes, chemically reactive molecules capable of binding to, among other things, DNA, providing a potential mechanism for light-induced A2E cytotoxicity.25
Knock-out mice lacking two of the genes controlling major proteins in the visual cycle (ABCA4 [the Stargardt's gene] and all-trans-retinal dehydrogenase 8 [RDH8], one of the main enzymes responsible for reduction of all-trans-retinal to the much less reactive all-trans-retinol) developed severe RPE/photoreceptor dystrophy at an early age and all of the salient features of AMD (lipofuscin deposits, drusen formation, basal laminar deposits, Bruch's membrane thickening and choroidal neovascularization).26 Exposure of these mice to light increased the severity of retinal damage, providing a scenario in which the accumulation of toxic visual cycle byproducts can cause cell damage and the chronic inflammatory stimulus that initiates clinical signs of AMD.26
Let's return to the brain. Following radiation exposure, the gradual loss of damaged vascular endothelial cells has been proposed as the chronic inflammatory stimulus. In the retina, the accumulation of toxic visual cycle derivatives, perhaps in combination with an underlying genetic defect, may produce a chronic source of damaged or dying cells that initiate inflammatory signaling and generate extracellular deposits that attract the attention of the complement system.
Treating Chronic Conditions
A chronic disease requires chronic treatment. Anti-inflammatory therapies minimize symptoms, but they may not address the root cause of the chronic inflammation, which is often unknown. Curative treatment may require biological rather than pharmacological approaches (i.e., cell replacement therapy or stem cell therapy).
In the Age-related Eye Disease Study, daily supplements of high doses of antioxidants and/or zinc for a period of five years delayed the rate of visual acuity loss in patients with intermediate or advanced AMD.27 Advanced age is an established risk factor for AMD, yet Stargardt's disease patients and the mouse models with visual cycle gene knockouts develop many of the characteristic features of AMD at an early age. Long-term, progressive oxidative damage to mitochondria has been suggested as a source of reactive oxygen species and as an underlying factor in a mitochondria-based model of AMD.28 These seemingly unrelated observations illustrate the complex interactions of visual cycle byproducts, oxygen, genetics, time and immune system activity that may all lead to retinal damage, but via different pathways.
In the mouse ABCA4 knockout model, treatment with Accutane, a vitamin A analog and inhibitor of rhodopsin regeneration, suppressed the formation of A2E epoxides.25 Retinylamine, another vitamin A analog, attenuated the light-induced damage in the ABCA4/RDH8 double-knockout model.26 In the ABCA4 knockout model of Stargardt's disease, drugs that specifically bind to RPE65, one of the proteins (and perhaps the rate-limiting step) in the visual cycle abolished the formation of A2E.29 Recent reports demonstrate that inhibitors of the visual cycle may represent promising approaches to treating AMD.30
Dr. Abelson, an associate clinical professor of ophthalmology at Harvard Medical School and senior clinical scientist at Schepens Eye Research Institute, consults in ophthalmic pharmaceuticals. Dr. Coderre is a medical writer at Ora Inc. in Andover.
1. Murphy HS. Inflammation. In: R. Rubin and D. Strayer, Eds. Rubin's Pathology: Clinicopathologic Foundations of Medicine. Baltimore: Lippincott Williams & Wilkins 2007:37.
2. Barcellos-Hoff MH. How tissues respond to damage at the cellular level: Orchestration by transforming growth . BJR Suppl 2005;27:123-7. b factor-
3. Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK. Physicochemical modeling of cell signaling pathways. Nat Cell Biol 2006;8:1195-203.
4. Vodovotz Y, Csete M, Bartels J, et al. Translational systems biology of inflammation. PLoS Comput Biol 2008;4.
5. Serhan CN and Savill J. Resolution of inflammation: The beginning programs the end. Nat Immunol 2005;6:1191-7.
6. Rubin P, Johnston CJ, Williams JP, et al. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys 1995;33:99-109.
7. Chiang CS, Hong JH, Stalder A, et al. Delayed molecular responses to brain irradiation. Int J Radiat Biol 1997;72:45-53.
8. Hauer-Jensen 1 gene b M, Richter KK, Wang J, et al. Changes in transforming growth factor- expression and immunoreactivity levels during development of chronic radiation enteropathy. Radiat Res 1998;150:673-80.
9. Williams JP, Hernady E, Johnston CJ, et al. Effect of administration of lovastatin on the development of late pulmonary effects after whole-lung irradiation in a murine model. Radiat Res 2004;161:560-7.
10. Diomede L, Albani D, Sottocorno M, et al. In vivo anti-inflammatory effect of statins is mediated by nonsterol mevalonate products. Arterioscler Thromb Vasc Biol 2001;21:1327-32.
11. Denham JW and Hauer-Jensen M. The radiotherapeutic injury--a complex 'wound'. Radiother Oncol 2002;63:129-45.
12. Hopewell J, Withers HR. Proposition: Long-term changes in irradiated tissues are due principally to vascular damage. Med Phys 1998;25:2265-8.
13. Lyubimova N and Hopewell JW. Experimental evidence to support the hypothesis that damage to vascular endothelium plays the primary role in the development of late radiation-induced CNS injury. Br J Radiol 2004;77:488-92.
14. Hobson B and Denekamp J. Endothelial proliferation in tumours and normal tissues: Continuous labelling studies. Br J Cancer 1984;49:405-13.
15. Li YQ, Chen P, Haimovitz-Friedman A, et al. Endothelial apoptosis initiates acute blood-brain barrier disruption after ionizing radiation. Cancer Res 2003;63:5950-56.
16. Hageman GS, Luthert PJ, Victor Chong NH, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res 2001;20:705-32.
17. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 2002;134:411-31.
18. Bok D. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc Natl Acad Sci U S A 2005;102:7053-54.
19. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA 2005;102:7227-32.
20. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005;308:419-21.
21. Spencer KL, Hauser MA, Olson LM, et al. Protective effect of complement factor B and complement component 2 variants in age-related macular degeneration. Hum Mol Genet 2007;16:1986-92.
22. Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med 2007;357:553.
23. Wang AL, Lukas TJ, Yuan M, et al. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS ONE 2009;4:e4160.
24. Weng J, Mata NL, Azarian SM, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell 1999;98:13-23.
25. Radu RA, Mata NL, Bagla A, and Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt's macular degeneration. Proc Natl Acad Sci USA 2004;101:5928-33.
26. Maeda A, Maeda T, Golczak M, Palczewski K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem 2008;283:26684-93.
27. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001;119:1417-36.
28. Liang FQ and Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res 2003;76:397-403.
29. Maiti P, Kong J, Kim SR, et al. Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry 2006;45:852-60.
30. Golczak M, Maeda A, Bereta G, et al. Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J Biol Chem 2008;283:9543-54.



