
Presentation
A 21-year-old African-American male presented to his general ophthalmologist because of decreased vision in both eyes. His symptoms began as photopsias and small black floaters in the right eye one month prior to presentation, followed by similar symptoms in the left eye about one week later. Soon after the floaters started in each eye, he had progressive visual loss in both eyes. The patient denied any other past ocular history.
Medical History
The patient's past medical history was significant for schizoaffective disorder. His medications included Depakote, Risperdal and Effexor. He had an allergy to penicillin. He denied any history of sickle cell disease or sarcoidosis. He also denied smoking or using alcohol or illicit drugs. Review of systems was negative.
Examination
Office examination revealed an uncorrected visual acuity of count fingers at 3 feet in the right eye and count fingers at 1 foot in the left eye, with no pinhole improvement. Pupillary reactions were brisk in each eye with no relative afferent defect. Extraocular motility was full in both eyes. Intraocular pressures were 17 mmHg in the right eye and 16 mmHg in the left eye. Given the patient's overall condition, he was unable to perform confrontational field or color vision testing. Slit-lamp biomicroscopy revealed 1+ and 2+ white cells in the anterior chamber of the right and left eye, respectively. He had minimal vitritis in both eyes. His dilated fundus examination was remarkable for many yellow-white choroidal lesions in the macula and periphery of both eyes. At that time, the patient was promptly referred to the retina clinic, where fundus photographs were taken (See Figure 1).
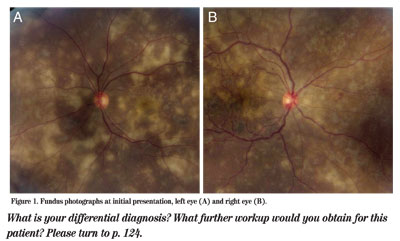
Figure 1. Fundus photographs at initial presentation, left eye (A) and right eye (B).
Diagnosis and Workup
Given the severity of the patient's visual loss in both eyes, he was admitted to the hospital. He was started on atropine 1% in both eyes twice a day and Pred Forte 1% in both eyes every hour. Although he was thought to have an inflammatory disease, a prompt infectious disease workup was performed. The following blood work was ordered to investigate infectious and inflammatory etiologies: complete blood count; basic metabolic panel; liver function panel; HIV testing; reactive plasma reagent; fluorescent treponemal antibody absorption test; toxoplasma titers; lyme titers; anti-nuclear antibody; erythrocyte sedimentation rate; blood cultures; purified protein derivative; angiotensin converting enzyme; and a chest X-ray. Aside from a slightly elevated blood glucose, the entire workup was negative. Given these results and the clinical findings, the condition was thought to be relentless placoid chorioretinitis (also referred to as ampiginous chorioretinopathy), one of the recently characterized variants of a white dot syndrome. He was started on prednisone 80 mg
At the three week follow-up visit, the patient's VA was unchanged. The anterior chamber and vitreous had no active inflammation, and the inflammatory lesions in the posterior pole had undergone pigmentation. At that time, a taper of the oral and topical steroids was started.
At the six-week follow-up visit, the patient's vision improved slightly to 20/200 in the right eye and 20/400 in the left eye. He is now being followed in the outpatient clinic and has been referred to a rheumatologist for consideration of chronic immunosuppression with a steroid-sparing agent.
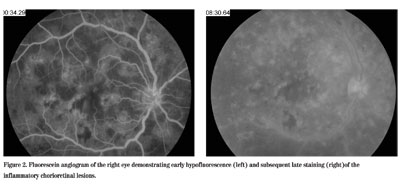
Figure 2. Fluorescein angiogram of the right eye demonstrating early hypofluoroescence (left) and subsequent late staining (right) of the inflammatory chorioretinal lesions.
Discussion
Bilateral multifocal inflammatory lesions have a broad differential diagnosis, including infectious, inflammatory and infiltrative disorders. Infectious etiologies to consider include syphilis, herpetic infections, cytomegalovirus, tuberculosis, lyme disease, candida, pneumocystis, endogenous endophthalmitis, histoplasmosis, toxoplasmosis and cystocercosis. Inflammatory disorders with this presentation can include sarcoidosis, white dot syndromes, Vogt-Koyanagi-Harada syndrome and Behçet's disease. Infiltrative processes include intraocular lymphoma and leukemia.
The white dot syndromes are characterized by multiple, yellow-white inflammatory lesions located in the choroid, retinal pigment epithelium and outer retina. They include serpiginous choroiditis, acute posterior multifocal placoid pigment epitheliopathy (AMPPE), multifocal choroiditis and panuveitis (MCP), multiple evanescent white dot syndrome (MEWDS), birdshot chorioretinopathy and punctate inner choroidopathy (PIC). The etiology of these syndromes is still not completely understood, and they are categorized by clinically descriptive names. Whether some of these conditions represent a spectrum of closely related diseases or a group of distinct clinical entities remains controversial.
Although the white dot syndromes are well-classified inflammatory diseases, many patients do not fit into the classic demographic or clinical presentation. In many cases, making the diagnosis of a white dot syndrome is difficult and occurs only after an extensive workup ruling out other infectious and inflammatory conditions. Our patient was diagnosed with relentless placoid chorioretinitis (ampiginous chorioretinopathy), a clinical entity first described in 2000, which has features that resemble both AMPPE and serpiginous choroiditis.
AMPPE is a bilateral disorder of young adults with acute, bilateral onset and mild visual loss, sometimes associated with a flu-like prodrome, cerebral vasculitis, thyroiditis, and/or erythema nodosum. It is usually monophasic and self-limited with good visual recovery. The retinal exam shows multiple, large, round, yellowish lesions primarily in the posterior pole that exhibit early hypofluorescence and late staining on fluorescein angiography. Serpiginous choroiditis is a bilateral disorder seen in young to middle-aged adults with an insidious, asynchronous onset. This condition is often recurrent months to years later and is associated with significant visual loss if the fovea becomes involved. The retinal exam demonstrates irregular, geographic, serpentine-like lesions that often emanate from the optic disc. On fluorescein angiography, these lesions exhibit early blockage and late staining at the active borders. Treatment usually consists of an immunosuppressive drug. Since our patient had characteristics of both disorders, a diagnosis of relentless placoid chorioretinitis was made.
In the original description of this disorder, patients had widespread lesions in the macula as well as the mid and far periphery, unlike typical AMPPE or serpiginous choroiditis. Many of the lesions progressed to chorioretinal atrophy within weeks. Despite this early apparent resolution, many new areas of inflammation can rapidly appear, often leaving many lesions scattered throughout the retina. These patients may experience severe visual loss. The best treatment has yet to be determined, but some patients appear to benefit from immunosuppressive therapy. Ultimately, it remains unclear whether relentless placoid chorioretinitis is a distinct clinical entity or a condition that falls into the spectrum of AMPPE and serpiginous choroiditis.
Dr. Baker is a first-year resident at Wills and would like to thank Dr. Robert Lehman for the case referral.


