
Presentation
A 47-year-old female of Greek origin presented to the Wills Eye emergency room with a one-month history of slowly progressive blurring of her vision in the left eye. Her symptoms were most prominent while reading or using the computer. She denied previous trauma or previous episodes of vision loss.
Medical History
The patient denied any previous medical or ocular history or any medication use. She had had a recent physical examination, which had been unremarkable, and blood work, including creatinine and BUN, which was within normal limits. She was unsure of her ophthalmic family history.
Examination
Her examination in the Wills Eye emergency room revealed a corrected visual acuity of 20/25 OD and 20/80 OS with no improvement with pinhole. Her pupils were round, equal, briskly reactive and without a relative afferent pupillary defect. Extraocular motility and confrontational visual fields were full in both eyes. Intraocular pressures were 15 mmHg OD and 12 mmHg OS by applanation tonometry. Amsler grid testing was within normal limits OD, but she noted central metamorphopsia OS. Color plates were full in both eyes. Slit-lamp examination of the lids, conjunctiva and anterior segment were within normal limits in both eyes except for trace nuclear sclerotic cataracts bilaterally.
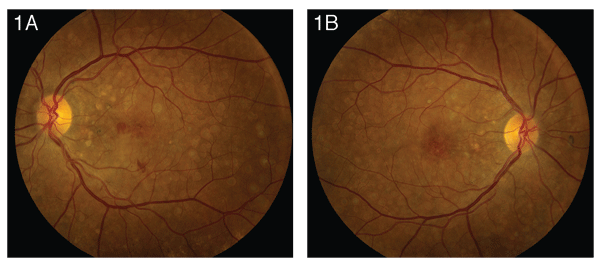
Dilated fundus exam of the left eye (See Figure 1a) revealed clear media with a healthy-appearing disc and vessels. Numerous sharply defined, round, white subretinal lesions were noted throughout the posterior pole, extending nasally to the disc. In the macula, these lesions were confluent. The retinal pigment epithelium also appeared slightly atrophic. A blunted foveal reflex and subretinal hemorrhage were also noted. Dilated fundus exam of the right eye (See Figure 1b) revealed symmetric findings, although the foveal reflex on the right was sharp. Dilated fundus exam of the patient's 18-year-old daughter was within normal limits. No other family members were available for examination.
Diagnosis and Workup and Treatment
Fluorescein angiography was performed and revealed normal choroidal flush and arterial filling with areas of staining hyperfluoresence consistent with drusen. Leaking hyperfluoresence in the macula consistent with an occult choroidal neovascular membrane was also seen (See Figures 2a and 2b).
The arrangement, appearance and symmetry of the drusen in this patient are consistent with dominant drusen syndrome. The FA revealed an occult CNVM in the left eye that was treated with intravitreal bevacizumab. Upon follow-up six weeks later, the patient's vision in the left eye had improved to 20/50.
Discussion
The differential diagnosis of degenerative drusen includes exudative age-related macular degeneration and membranoproliferative glomerulonephritis type II. Inherited drusen syndromes also have this general fundus appearance. Retinal "fleck" disorders, including fundus flavimaculatus, fundus albipunctatus, multifocal best disease and fleck retina with congenital hemeralopia, are also included in the differential. White flecks may be seen in white-dot syndromes and disseminated choroiditis. 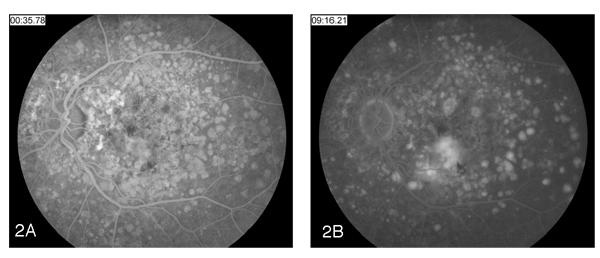
Robert Doyne described dominant drusen in 1899 in patients living in
Dominant drusen can be distinguished from typical AMD by fundus appearance. In dominant drusen, the drusen cover the posterior pole, extending nasally to the disc, often in a radial, mosaic or honeycomb pattern. The lesions closer to the fovea tend to be larger and coalesce with age. The appearance is often symmetric between the two eyes.
The drusen are often visible by ophthalmoscopy between ages 20 and 30, although the patient with dominant drusen syndrome usually does not present with symptoms until after age 40. The most common presenting symptoms are painless decrease in vision and metamorphopsia. As in this case, the patient's symptoms may also be secondary to a CNVM.
Ancillary testing may be helpful in the diagnosis of dominant drusen. Fluorescein angiography reveals multiple sharply defined hyperfluorescent lesions that do not perfectly correspond to the ophthalmoscopic appearance. Larger drusen and those located around the disc show autofluorescence. Optical coherence tomography shows thickening of the RPE-Bruch's membrane complex with preservation of the neurosensory retina. Electroretinography and electro-oculography are within normal limits except in advanced cases.
A single missense mutation (arg345trp) on exon 10 in the EFEMP1 gene of patients with dominant drusen results in a fibrillin-like extracellular matrix protein expressed in the retinal pigment epithelium and retina. This mutation is found in both Malattia Leventinese and Doyne's honeycomb dystrophy. Phenotypic variation of family members with the EFEMP1 mutation is also significant. These factors have led to the determination that other genetic and environmental modulators are important in the pathogenesis. Individuals with clinical dominant drusen but without the genetic mutation have been identified.
The long-term prognosis in this case is unknown, as intravitreal bevacizumab for treatment of CNVM in dominant drusen has not, to our knowledge, been previously reported.
Dr. Ehrlich would like to thank
Evans K et al. Assessment of the Phenotypic Range Seen in Doyne Honeycomb Retinal Dystrophy. Arch Ophthalmol 1997;115: 904-10.
Matsumoto M, Traboulsi EI. Dominant radial drusen and Arg345Trp EFEMP1 mutation. Am J Ophthalmol 2001;131:810-2.
Yi C, Pan X, Russell S, Agarwal A, Sternberg P Jr. Diagnostic and Therapeutic Challenges. Retina 2006;26:688-92.
Narendran N, Guymer RH, Cain M, Baird PN. Analysis of the EFEMP1 gene in individuals and families with early onset drusen. Eye 2005;19(1):11-5.
Pager CK, et al. Malattia leventinese presenting with subretinal neovascular membrane and hemorrhage. Am J Ophthalmol 2001;131:517-8.
Souied EH. Leveziel N. Letien V. Darmon J. Coscas G. Soubrane G. Optical coherent tomography features of malattia leventinese. Am J Ophthalmol 2006;141:404-7.
Souied EH. Leveziel N. Querques G. Darmon J. Coscas G. Soubrane G. Indocyanine green angiography features of Malattia leventinese. Br J Ophthalmol 2006;90(3):296-300.
Stone EM, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nature Genetics 1999;22:199-202.


