Olfatt Shaker, MD, Shaker A. Mousa, PhD, MBA, FACC, FACB
Retinitis pigmentosa is a slowly progressing hereditary disorder in which abnormalities of the photoreceptors (rods and cones) or the retinal pigment epithelium of the retina lead to progressive visual loss.1 The name retinitis pigmentosa was first applied by Dutch ophthalmologist Franziscus C. Donders in 1857. That is the phenotypic description of several related, yet distinct, dystrophies of the photoreceptors and the pigment epithelium. It affects about one in every 4,000 people in the United States.2
RP is characterized by multiple mutations in the genes of the retina and the RPE. It is an accumulation of a group of hereditary mutations that lead to progressive dysfunction, cell loss and atrophy of the retinal tissues. The cell atrophy spreads to most layers of the retina, resulting in visual impairment. Eventually, there is a widespread atrophy of several if not most layers of retina which ultimately leads to a loss of central vision dysfunction and complete loss of vision. RP affects males more than females. Though it is frequently diagnosed during childhood, some people do not have noticeable symptoms until later in life. Age of onset varies in different types of RP from infancy to adulthood with an average age of diagnosed patients with RP of about 35.1 years.3,4
People with RP usually first notice difficulty seeing in dim lighting and gradually lose peripheral vision. The loss of vision may be subtle and unnoticed by the patient, or severe and result in profound loss of peripheral visual field resulting in tunnel vision or even worse. RP is characterized by constriction of the visual fields, night blindness, fundus changes, including "bone corpuscle," which are lumps of pigment.
Retinitis Pigmentosa Classification
There are various ways in which the disease is subdivided and classified. One is according to the mode of inheritance: autosomal dominant RP (adRP), autosomal recessive RP (arRP) and X-linked recessive RP (xlRP), which is the most severe form. Other ways of classification vary according to retinal involvement and fundus appearance, age of onset or molecular defects.
Furthermore, the xlRP can be either recessive, affecting males only, or dominant, affecting both males and females; females are always more mildly affected. Some digenic and mitochondrial forms have also been described: X-linked (5 to 15 percent), autosomal dominant (30 to 40 percent) and the remainder assumed autosomal recessive (50 to 60 percent). RP unassociated with other abnormalities is inherited most frequently (50 to 60 percent) as an autosomal recessive, next as an autosomal dominant (30 to 40 percent) and least frequently (5 to 15 percent) as an X-linked recessive in the white
Cell biology and molecular genetics have led to the enhancement in understanding of possible cause and the root problem of this disorder. Histopathological studies have revealed that initially there are photoreceptors abnormalities that are caused by genetic mutation that leads to abnormal apoptosis pathway, although the reason this apoptosis turns on is unclear. These findings have resulted in better understanding of the disease along with current and future treatment options.6,7
Clinical Manifestations
Night blindness is a hallmark symptom of RP and usually occurs during the first two decades of life, with a median age of onset being 10.7 years in arRP and 23.4 years in adRP.8 Visual field loss is the second hallmark feature of RP as a result of progressive loss of peripheral cell function, leading to peripheral vision loss. Field deficits are the first of symptoms recognized and identified by many RP patients; there also tends to be a drastic change in their superior visual field. Generally the loss of visual field is symmetrical in both eyes with the exception of xlRP, where there are asymmetrical visual field changes and loss of vision in both eyes. The loss of visual field is still around the fundus with greater loss in the eye with more pigment abnormalities.9 Moreover, patients with adRP are more likely to retain good visual acuity past 60 years of age than the patients with arRP or xlRP.
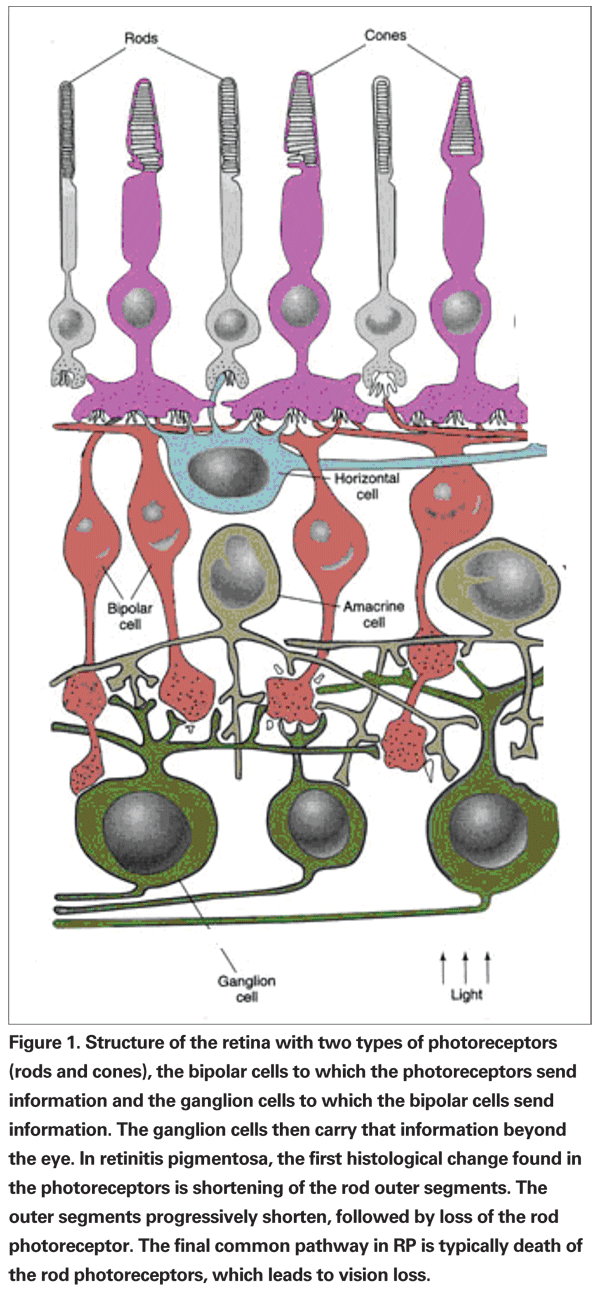
In addition, patients may not even feel peripheral vision loss if central vision remains clear until vision is decreased to tunnel vision, when the change is dramatic and it is realized by the patient. Moreover, RP patients could experience central vision loss even before any serious peripheral vision loss. Color vision in patients with RP usually remains good until visual acuity is worse than 20/40; however, patients experiencing central vision loss could have impaired color vision even before any extensive peripheral vision loss.10 During the process of declining vision, RP patients experience photopsia, the sensation of tiny blinking or shimmering of light. This sometimes could be confused with ophthalmic migraines, which could have similar type of symptoms.
Mechanism of Retinal Degeneration
Photoreceptor abnormalities have been attributed to abnormal rhodopsin mutation and accumulation that leads to death in photoreceptor cells. This is followed by RPE detachment and cell accumulation causing bone-spicule pigmentation.11 There are reactive changes in all cell types subsequent to photoreceptor cell death, which further damage the remaining part of the retina and lead to further progression of the disease. Retinal vascular abnormalities and vascular attenuation lead to decreased blood flow to the retina in advanced RP.11
Numerous mutation loci linked to RP have been found for different RP types. Rhodopsin is one of the major proteins of the photoreceptor cells and also one of the most common proteins to undergo mutation in RP.13 Overall, many diverse genes could be implicated in RP. They could either be specific for the eye, which could include the visual transduction cascade, structural proteins, retinoid cycle, outer segment renewal and transcription factors, or they could be widely expressed throughout the body such as splicing factors and nucleotide metabolism.14
However, the molecular mechanisms underlying the different forms of RP are not known, due to the genetic and functional variability of the involved proteins. One common thing in all RP types is photoreceptor cell death via the apoptosis attributed to different mechanisms. One of the possible mechanisms is believed to be the activation of caspase-12 and AIF (apoptosis inducing factor).15,16 In vivo and in vitro studies showed that AIF and caspase 12 are activated and localized together with fragmented chromatin in apoptotic photoreceptors.17 This could be due to oxidative stress on the ER leading to the activation of the apoptotic pathway. There is also the involvement of calpain and its function in the apoptotic cascade.17 Calpains do not directly cause chromatin condensation but they are proteases activating apoptotic factors, such as caspase 12 and AIF.17 Calpains are engaged by high calcium and ER stress and are probably upstream to both AIF and caspase 12 activation, leading to apoptosis.18 Activation of calpain and cathepsin D has been associated with cell death in RP.19,20
Diagnosis and Testing
The diagnosis of RP relies upon documentation of progressive loss in photoreceptor function by electroretinography and visual field testing. The mode of inheritance of RP is determined by family history. There is some evidence that RP could be detected in early life by ERG testing. There is a delay in the amplitude of ERG in patients with early disease, and it becomes smaller as the disease progresses. This could detect asymptomatic patients with RP, years before they show abnormalities in routine ocular examination. In a study, individuals age 6 and older with normal ERGs and a family history of retinitis pigmentosa have not been observed to develop retinitis pigmentosa at a later time.21,22
Attenuated retinal vessels, mottling and granularity of the retinal pigment epithelium, optic nerve head pallor and bone spicule intraretinal pigmentation are classically seen in an RP patient's fundus. In advanced disease, RPE atrophy and choriocapillaris lead to pallor fundus and visibly larger choroidal vessels. Furthermore, during the late stages of the disease the retinal vessels appear thread-like due to severe vessel attenuation. Retinal attenuation and fine mottling or granularity of the RPE is one of the earliest signs to be found in RP patients with fundus abnormalities. The diagnosis should be confirmed by electrophysiology, and genetic counseling should be undertaken, as well as low-vision service delivery. Other family members should be examined, particularly the mother of a male with RP who may have an X-linked inheritance pattern.23,24
Differential Diagnosis
Individuals who present with initial symptoms of photopsia, abnormal central vision, abnormal color vision or marked asymmetry in ocular involvement may not have RP, but another retinal degeneration or retinal disease. Some disorders to consider in the differential diagnosis of typical RP: Usher syndrome, chorioderemia, gyrate atrophy of the choroid and retina, cone-rod dystrophy, Leber's congenital amaurosis, Retinal-renal Senior-Loken syndrome and mitochondrial disorders.25,30
Management of RP
Treatment of manifestations:
• Retinal degeneration. Therapy with 15,000 IU per day of vitamin A palmitate supplements is suggested to delay the progression of RP. Daily supplements of this dosage of vitamin A have been shown to slower the rate of progression of RP in a randomized, controlled trial, with the course of retinal degeneration monitored by ERG.31,32 In contrast, the use of vitamin E 400 IU per day supplements was found to increase the rate of progression of RP. A possible reason for this could be reduced amounts of vitamin A reaching the eye with concomitant use with vitamin E.33
• Cataracts. Most affected individuals with a visual field of greater than 10 degrees are not incapacitated by posterior subcapsular cataracts. Those with a visual field of less than 10 usually report significant improvement in visual function following lens extraction.34
• Optical aids. Use of CPF 550 lenses (Corning Photochromatic Filter, manufactured by Corning Glass Works), which filter out 97 to 99 percent of the spectral and ultraviolet energy below 550 nm wavelength, has been promoted for individuals with RP to increase eye comfort by reducing glare and internal light scatter and to reduce adaptation time from light to dark and vice versa.
Therapies Under Investigation
The use of docosahexaenoic acid (DHA) 1,200 mg per day supplement in addition to 15,000 IU per day vitamin A supplement didn't show any clear benefit to patients suffering from RP in two different studies. One involved 44 xlRP patients and the other 208 patients with various types of RP. Both studies, however, showed that patients with the highest DHA concentration in red blood cells had the slowest rate of degeneration.35,36 In addition, further analysis within the subjects showed that participants receiving at least 1,200 mg per day of omega-3 fatty acids (DHA) with vitamin A 15,000 IU per day for two years had a slower disease progression than those consuming vitamin A 15,000 IU per day with less or no DHA supplement.36,37 So, the use of a DHA supplement 1,200 mg per day along with vitamin A could be of potential benefit to the patients with RP in slowing the disease's progression. Furthermore, vitamin A supplements were safe in 146 patients with RP between 18 and 54 years of age who took 15,000 IU per day of vitamin A for less than or equal to 12 years. There were no signs of liver toxicity or other adverse effects associated with it.38
Calcium channel blockers such as diltiazem were shown to reduce the rate of progression of RP in an animal study; however, subsequent studies have failed to prove any benefit. Until more data is available on the benefit of diltiazem or other calcium channel blockers, their use in preventing progression in RP is not recommended.39
In 2001, a study of oral supplementation with 20 mg per day of lutein for six months demonstrated increased macular pigment in approximately 50 percent of individuals with RP or Usher syndrome, but no change in central vision.40 The long-term effects of such supplementation are unknown.
Future Directions
Efforts are being invested to identify the involved genes, to understand the pathophysiologic mechanisms and to find efficient therapeutic strategies.
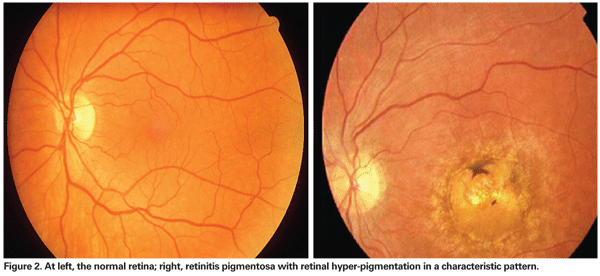
• Gene therapy. Knowledge of the genetic basis of many severe ocular diseases may allow for alternative treatments by gene therapy. XIAP (X-linked inhibitors of apoptosis) gene therapy is known to prevent apoptosis in ischemic retina.41 This therapy has restored both functional and structural protection to the retina. Its use in RP patients also showed some potential in preventing photoreceptor cell death. Apoptosis is considered the final and common step for all types of RP patients regardless of the mutations in the genes. The use of XIAP to prevent cell death in retinal cells could result in prevention of RP41; however, more studies are required for this theory.
One animal study showed mixed response to this technique in prevention of RP in rat models. It was able to prevent apoptosis in P23H animal models but showed no response in S334ter animals. More analysis of this technique in animals is required to ensure the effectiveness of XIAP gene therapy in preventing photoreceptor cell death.42
• Artificial silicon retina microchip implant. The ASR microchip, containing microelectrodes with microphotodiodes powered by incident light rays, is placed in the subretinal space. It was found to be beneficial and functional in patients more than two years postop, with no signs of rejection of the implant. There were visual improvements which included better perception of brightness, contrast, color, movement, shape, resolution and visual field size. In addition, the visual improvement was not restricted to the area of implant but was also visible far away from the implant in other parts of retina, suggesting some possible neurotrophic protection associated with it. Certain procedures such as high-powered ultrasound should be avoided in implanted eyes. The use of implant in treating RP, however, requires more trials and research beyond the Phase I clinical trial to determine its optimal use and benefit.43,44
• Encapsulated cell technology implant. The ECT implant is genetically engineered to deliver specific ciliary neurotrophic growth factor (CNTF) into the eye. This polymer implant contains genetically modified human RPE cells, which secrete CNTF into the vitreous of the patients' eyes. This technology enables the controlled, continuous and long-term delivery of therapeutic proteins directly to the back of the eye. During the Phase I safety analysis, the implant was put into one eye of 10 patients with RP and removed after six months. There were no serious adverse effects reported during the implant period, such as retinal detachment, intraocular pressure increase, infection or serious inflammation.45-48 There was, however, a single instance of shallow choroidal detachment in one eye receiving the lower-dose device. This same eye also developed a nuclear cataract 4.5 months after the implant removal. It could possibly be due to the mechanical insults to the area related to the surgeries rather than to effects of CNTF or the implant. The implant was well-tolerated and showed an improvement in visual acuity scores of some patients. This delivery system is encouraging, and the system is now being tested for a Phase II/III trial. However, long-term safety and efficacy of the implant cannot be determined until further data is available.45-48
• Stem cell therapy. Results of stem cell research has been uncertain and very little is known about it. There are many diseases that are related to cell degeneration that could potentially benefit from stem cell therapy. Furthermore, many researchers are working on finding ways to control the use of stem cells. Stem cells when used appropriately could be of great benefit and could lead to a possible cure for many diseases. However, the use of stem cells has its own risks and disadvantages.49-51
Different types of stem cells have different properties associated with them. Embryonic stem (ES) cells could potentially be differentiated into any cell of the body; however, it is not fully known how to govern the differentiation of cells, and more importantly, when to stop them to prevent possible unwanted results. These cell types also have surface antigen, which could lead to immune rejection by the host organism. To prevent rejection in the host subject, cloning of ES cells is possible but that opens the door to even more complications, as cloning could result in mutations and be prone to tumors.52
Progenitor stem cells, on the other hand, are easier to control and handle and also do not have the issue of host rejection. They differentiate into a limited number of cells. In the retina, the idea is that these cells would integrate into the damaged retina and protect it from further damage and also could help in regeneration of the lost cells. Progenitor cells have similar complications to ES cells in that so little is known about them and their properties.53-55
Although there is progress in this field, more animal data is needed before any conclusions could be made on stem cell research. There have been cases of human stem cell trials which could be beneficial, but could also turn out to be disastrous for the patients. A Phase I study looking at the viability of bone marrow stem cells in RP patients in India is one such ongoing human trials.56 However, very careful observation is required for such trials especially if the trial is only being conducted in one country, while the compound being studied is produced in another. Overall, stem cell research has a very bright future especially in degenerative diseases such as RP, but it is very premature to come to any conclusions based on the little data available.57
• Areas of exploratory research. In a prospective, comparative control study involving 16 RP patients, aqueous humor from the subjects was collected to determine the concentration of vascular endothelial growth factor A, or VEGF-A. VEGF-A was found in very low concentrations in the aqueous humor of RP patients when compared to the control group.58 VEGF-A is an important growth factor for angiogenesis and vasculature growth. Decrease in VEGF-A's levels could cause the lack of angiogenic actions attributed to RP and might possibly explain some of the clinical manifestations of this disease, such as narrowing and fibrotic degeneration of retinal blood vessels. Further testing with VEGF may prove to prevent the loss of blood supply, which could potentially lead to retinal cell survival. Again, more analysis is required as VEGF is also the factor which leads to age-related macular degeneration; hence its viability should be considered against the potential risks.
In a recent trial, 10 patients received retinal implants consisting of neural retinal progenitor cell layers (sheets) with its RPE in retinitis pigmentosa and dry age-related macular degeneration patients with 20/200 or worse vision in the surgery eye. There was some improvement noted in visual acuity in about seven of 10 patients with three of them being RP patients. This experiment is promising for the future of retinal diseases; however, more trials are needed to ensure the effectiveness and safety of this method in treating RP. Since this trial had both AMD and RP patients, a trial with only the various RP patients would be better in delineating the benefits of this technique to restore vision in RP patients.59
Although RP is a hereditary disorder that is not fully understood and doesn't have any cure, advances toward its treatment are promising, especially the use of implants and various growth factors. The use of stem cells could also prove to be a landmark as they are thought to have potential to re-grow the damaged parts in the retina. Much research is ongoing in understanding the proper etiology of this disease at a molecular level.
The authors are with The Pharmaceutical Research Institute at the Albany College of Pharmacy and Health Sciences,
1. Delyfer MN, Leveillard T, Mohand-Said S, Hicks D, Picaud S, Sahel JA. Inherited retinal degeneration: therapeutic aspects. Biol Cell 2004;96:261–69.
2. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368:1795–809.
3. Tsujikawa M, Wada Y, Sukegawa M, Sawa M, et al. Age at onset curves of retinitis pigmentosa. Arch Ophthalmol 2008;126: 337-40.
4. The Merck Manual of Diagnosis and Therapy 18th Edition. Beers MH, Berkow R, eds. Whitehouse Station, Merck Research Laboratories, 2006.
5. de Beus A, Small KW. Retinitis Pigmentosa. eMedicine. September, 2005.
6. Marmor MF, Aguirre G, Arden G, Berson E, Birch DG, Boughman JA et al. Retinitis pigmentosa. Ophthalmology 1983;90:126-31.
7. Pagon RA. Retinitis pigmentosa. Surv Ophthalmol 1988;33:137-77.
8. Tanino TO. Studies on pigmentary retinal dystrophy. Age of onset of subjective symptoms and the mode of inheritance.
9. Jacobson SG, Yagasaki K, Feuer WJ, Román AJ. Interocular asymmetry of visual function in heterozygote's of X-linked retinitis pigmentosa. Exp Eye Res 1989;48:679-91.
10.
11. Li ZY, Possin ED, Milam AH, Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology 1995;102:805-16.
12. Grunwald JE, Maguire AM, Dupont J, Retinal hemodynamic in retinitis pigmentosa. Am J Ophthalmol 1996;122:502-508.
13. Nathans J. Rhodopsin: Structure, function and genetics. Biochemistry 1992;31:4923-31.
14. Hartong DT, Berson EL, Dryja TB. Retinitis pigmentosa. Lancet 2006;368(9549):1795-809.
15. Wootz H, Hansson I, Korhonen L, Näpänkangas U, Lindholm D. Caspase-12 cleavage and increased oxidative stress during motoneuron degeneration in transgenic mouse model of ALS. Biochem Biophys Res Commun 2004;322:281-86.
16. Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. Embo J 2003;22:5435-45.
17. Sanges D, Comitato A, Tammaro R, Marigo V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci USA 2006;103:17366-71.
18. Marigo V. Programmed cell death in retinal degeneration: targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle 2007;6:652-55.
19. Paquet-Durand F, Azadi S, Hauck SM, Ueffing M, et al. Calpain is activated in degenerating photoreceptors in the rd1 mouse. J Neurochem 2006;96:802-14.
20. Doonan F, Donovan M, Cotter TG. Activation of multiple pathways during photoreceptor apoptosis in the rd mouse. Invest Ophthalmol Vis Sci 2005;46: 3530-38.
21. Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci 1993;34: 1659-76.
22. Berson EL, Rosner B, Sandberg MA, Weigel-DiFranco C, Dryja TP. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His). Arch Ophthalmol 1991;109:92-101
23. Reese BE, Harvey AR, Tan SS. Radial and tangential dispersion patterns in the mouse retina are cell-class specific. Proc Natl Acad Sci
24. Tan SS, Godinho L, Tam PP. Cell lineage analysis. X-inactivation mosaics. Methods Mol Biol 2000;135:289-95.
25. Plantinga RF, Pennings RJ,
26. Mears AJ, Hiriyanna S, Vervoort R, Yashar B, et al. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet 2000;67:1000-1003.
27. Koenekoop RK. RPGRIP1 is mutated in Leber congenital amaurosis: A mini-review. Ophthalmic Genet 2005;26:175-79.
28. Yang Z,
29. Otto EA, Loeys B, Khanna H, Hellemans J, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 2005;37:282-88.
30. MojonD. Eye diseases in mitochondrial encephalomyopathies. Ther Umsch 2001;58:49-55.
31. Berson EL, Rosner B, Sandberg MA, Hayes KC, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol 1993;111:761-72.
32. Massof RW, Finkelstein D. Supplemental vitamin A retards loss of ERG amplitude in retinitis pigmentosa. Arch Ophthalmol 1993;111:751-54.
33. Berson EL. Nutrition and retinal degenerations. Int Ophthalmol Clin 2000;40:93-111.
34. Jackson H, Garway-Heath D, Rosen P, Bird AC, Tuft SJ. Outcome of cataract surgery in patients with retinitis pigmentosa. Br J Ophthalmol 2001;85:936-38.
35. Berson EL, Rosner B, Sandberg MA, Weigel-DiFranco C, et al., Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol 2004;122: 1297-305.
37. Berson EL, Rosner B, Sandberg MA, Weigel-DiFranco C, et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch Ophthalmol 2004;122:1306-14.
38. Sibulesky L, Hayes KC, Pronczuk A, Weigel-DiFranco C, Rosner B, Berson EL. Safety of <7,500 RE (<25,000 IU) vitamin A daily in adults with retinitis pigmentosa. Am J Clin Nutr 1999;69:656-63.
39. Pearce-Kelling SE, Aleman TS, Nickle A, Laties AM, et al. Calcium channel blocker D-cis-diltiazem does not slow retinal degeneration in the PDE6B mutant rcd1 canine model of retinitis pigmentosa. Mol Vis 2001;7:42-7.
40. Aleman TS, Duncan JL, Bieber ML, de Castro E, et al. Macular pigment and lutein supplementation in retinitis pigmentosa and Usher syndrome. Invest Ophthalmol Vis Sci 2001;42:1873-81.
41. Renwick J, Narang MA, Coupland SG, Xuan JY, et al. XIAP-mediated neuroprotection in retinal ischemia. Gene Ther 2006;13:339-47.
42. Leonard KC, Petrin D, Coupland SG, Baker AN, et al. XIAP protection of photoreceptors in animal models of retinitis pigmentosa. PLoS ONE 2007;2:e314-e22.
43. Chow AY, Chow VY, Packo KH, Pollack JS, et al. The artificial silicon retina microchip for the treatment of vision loss from retinitis pigmentosa. Arch Ophthalmol 2004;122:460-69.
44. Chow AY, Chow VY, Packo KH, Pollack JS. The Artificial Silicon Retina Microchip for the Treatment of Retinitis Pigmentosa: 2 to 4 1/2 Year Update. Invest Ophthalmol Vis Sci 2005;46:1140.
45. Del Amo EM, Urtti A. Current and future ophthalmic drug delivery systems: A shift to the posterior segment. Drug Discov Today 2008;13:135-43.
46. Sieving PA, Caruso RC, Tao W, Coleman HR, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: Phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci USA 2006;103:3896-901.
47. Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin Biol Ther 2006;6:717-26.
48. Hossain P, Seetho IW, Browning AC, Amoaku WM. Artificial means for restoring vision. BMJ 2005;330(7481):30-3.
49. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998;282(5391):1145-47.
50. Pedersen RA. Embryonic stem cells for medicine. Sci Am 1999;280:68-73.
51. Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 2000;18:675-79.
52. Arnhold S, Klein H, Semkova I, Addicks K, Schraermeyer U. Neurally selected embryonic stem cells induce tumor formation after long-term survival following engraftment into the subretinal space. Invest Ophthalmol Vis Sci 2004;45:4251-55.
53. Tropepe V, Coles BL, Chiasson BJ, Horsford DJ, et al. Retinal stem cells in the adult mammalian eye. Science 2000;287(5460):2032-36.
54. Nishida A, Takahashi M, Tanihara H, Nakano I, et al. Incorporation and differentiation of hippocampus-derived neural stem cells transplanted in injured adult rat retina. Invest Ophthalmol Vis Sci 2000;41(13):4268-74.
55. Ahmad I, Tang L, Pham H. Identification of neural progenitors in the adult mammalian eye. Biochem Biophys Res Commun 2000;270:517-21.
56. Kumar AP, Tandon R, Kumar L, Mohanty S. Use of Autologous Bone Marrow Derived Stem Cells for Rehabilitation of Patients with Dry Age Related Macular Degeneration and Retinitis Pigmentosa: Phase-1 Clinical Trial. Indian J Med &
57. Mooney I, LaMotte L. A review of the potential to restore vision with stem cells. Clin Exp Optom 2008;91:78-84.
58. Salom D, Diaz-Llopis M, García-Delpech S, Udaondo P, et al. Aqueous Humor Levels of Vascular Endothelial Growth Factor in Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 2008;49:3499-502.
59. Radtke ND, Aramant RB, Petry HM, Green PT, et al. Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am J Ophthalmol 2008;146:172-182.



