The Food and Drug Admin-istration has approved Lucentis (rani- bizumab injection, Genentech/Roche) for the treatment of macular edema following retinal vein occlusion. The agency approved this new indication after a six-month priority review.
The BRAVO study assessed the safety and efficacy profile of Lucentis in a total of 397 patients with macular edema following branch-RVO. The CRUISE study assessed the safety and efficacy profile of Lucentis in a total of 392 patients with macular edema following central-RVO. During the first six-month period, participants in both trials received monthly injections of either 0.3 mg or 0.5 mg of Lucentis (n=527) or monthly sham injections (n=262). The primary endpoint of both studies was mean change from baseline in best-corrected visual acuity at six months compared with patients receiving sham injections. The studies were not designed to compare the two doses of Lucentis.
In the BRAVO study, the percentage of patients in the Lucentis 0.5-mg study arm who gained 15 or more letters in BCVA from baseline at month six was 61 percent (compared with 29 percent in the sham injection arm). In the CRUISE study, the percentage of patients in the Lucentis 0.5-mg study arm who gained 15 or more letters in BCVA from baseline at month six was 48 percent (compared with 17 percent in the sham injection arm). At month six, patients in BRAVO who received 0.5 mg of Lucentis had a mean gain of 18.3 letters (compared to 7.3 letters in patients receiving sham injections). In the CRUISE study, at month six, patients who received 0.5 mg of Lucentis had a mean gain of 14.9 letters (compared to 0.8 letters for patients receiving sham injections).
The adverse events reported in BRAVO and CRUISE were similar to previous studies, and no new safety events were observed. In BRAVO and CRUISE, the most common ocular adverse events that occurred in the Lucentis arms included conjunctival hemorrhage (48 percent) and eye pain (17 percent). Among non-ocular serious adverse events in the BRAVO study, one arterial thromboembolic event occurred in the sham injection group and two events occurred in the Lucentis 0.5-mg dose group: one cerebrovascular accident that resulted in death and one myocardial infarction. In CRUISE, non-ocular serious adverse events were uncommon and included one case of either myocardial infarction or acute coronary syndrome in each of the three groups. No cerebrovascular accidents or deaths occurred during the six-month treatment period in CRUISE.

QLT: Combo AMD Therapy Cuts Retreatments
QLT released final 24-month results from the Phase II RADICAL study (reduced fluence Visudyne anti-VEGF-dexamethasone in combination for AMD lesions) in patients with exudative age-related macular degeneration. The study aimed to determine if Visudyne combined with Lucentis reduced retreatment rates compared with Lucentis monotherapy, while main- taining similar vision outcomes and an acceptable safety profile. Three Visudyne-Lucentis combination therapies were evaluated against Lucentis monotherapy.
The results of the 24-month analysis are consistent with the primary analysis results after 12 months of follow-up. The 24-month results showed that significantly fewer retreatment visits were required with combination therapies than with Lucentis monotherapy. Mean visual acuity change from baseline was not statistically different among the treatment groups, although the sample sizes were insufficient to draw definitive conclusions regarding VA outcomes. Overall adverse event incidence was similar across treatment groups, with no unexpected safety findings.
Of the four treatment groups, the triple-therapy, half-fluence group had the fewest retreatment visits compared with Lucentis monotherapy. Through 24 months, patients in the triple-therapy, half-fluence group had a mean of 4.2 retreatment visits compared with 8.9 for patients who received Lucentis monotherapy (p<.001). In the second year, the average number of retreatment visits in the triple-therapy, half-fluence group (1.2) was approximately one-third that of the Lucentis monotherapy group (3.0). At the month 24 examination, mean VA in the triple therapy half-fluence group improved 1.8 letters fewer compared with the Lucentis monotherapy group (p=0.71). Mean retreatment visits and mean VA change from baseline for each treatment group are presented in the table below.
All results presented are based on per-protocol analyses. Patients were evaluated for VA and safety over the 24-month follow-up period. Patients were assessed to determine if retreatment was needed at each follow-up visit from month one (combination therapy groups) or month three (Lucentis monotherapy group), except for the final visit. Overall, 21 patients left the study early for reasons unrelated to Visudyne or Lucentis.
The results for secondary VA endpoints in the RADICAL study (percentage of patients with VA improvement ¡Ý15 letters, VA change ¡Ýzero letters and VA loss <15 letters) were consistent with the results for the primary VA endpoint (mean VA change from baseline). Ocular adverse events considered associated with treatment were reported for 30 to 38 percent of patients in the combination therapy groups, compared with 27 percent of patients in the Lucentis monotherapy group. The higher incidence of these events with combination therapy is primarily due to vision disturbance events, which are transient and known to be associated with Visudyne therapy.
The full results of the 24-month final analysis of the study will be presented at the 28th Annual Meeting of the American Society of Retina Specialists in August.
Positive Data For Iluvien DME Treatment Presented
Alimera Sciences announced that a new analysis of data from its Phase III (FAME) clinical trials for Iluvien, the company¡¯s investigational intravitreal insert being studied for diabetic macular edema, indicates that patient vision continues to im-prove significantly over 30 months.
The analysis focused on the primary efficacy variable of the number of patients who improved by 15 letters or more, based on observed cases. At month 24, 31 percent of the low-dose patients (p=0.003) had improved vision of 15 letters or more and at month 30, with a sample size of 123 patients, an improvement in visual acuity of 15 letters or more was seen in 40 percent of the patients (p=0.002). Statistical significance versus control was seen by three weeks among the observed cases, and this significance was maintained through month 30. The complete 36-month dataset will be presented after the trial concludes in October 2010.
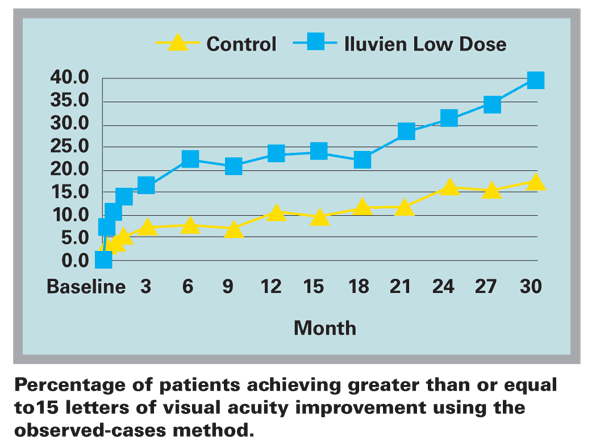
¡°While there have been some exciting developments of late in investigational DME therapies, this preliminary, 30-month observed cases data for Iluvien is of significant interest to retinal physicians,¡± said Barry Kuppermann, MD, PhD, professor and chief of the Retina Service at University of California, Irvine. ¡°For patients to continue to have vision improvement, at this level, up to 30 months after first receiving Iluvien is impressive.¡±
Alimera is currently conducting two Phase III pivotal clinical trials (collectively known as the FAME Study) for Iluvien involving 956 patients in sites across the United States, Canada, Europe and India to assess the efficacy and safety of Iluvien with two doses, a high and low dose, in the treatment of DME. In December 2009, the month 24 clinical readout from the FAME Study was completed and announced. Alimera plans to file a New Drug Application in the
Although both a low and high dose of Iluvien were studied in these trials, only the low dose is shown in Exhibit 1 as Alimera intends to commercialize this dose if approved by the FDA. In the previous dataset presented, data imputation employed the ¡°last observation carried forward¡± method, for data missing because of patients who discontinued the trial or were unavailable for follow-up. In the analysis presented today, no data imputation was employed and only ¡°observed cases¡± were considered in which only those patients for whom data was available at the clinical visit were analyzed.
B+L Acquires Sirion¡¯s Zirgan
Bausch + Lomb HAS acquired Zirgan (ganciclovir ophthalmic gel 0.15%) from Sirion Therapeutics. Zirgan was approved by the FDA in 2009 as a topical anti-viral for the treatment of acute herpetic keratitis.
Unlike older anti-viral drugs that affect both healthy and infected cells, Zirgan selectively targets the replication of herpes simplex virus DNA. Sirion introduced Zirgan to the
The FDA has designated Zirgan as an orphan drug, conveying special status for rare diseases or conditions that affect fewer than 200,000 patients in the
Zirgan is available by prescription through


