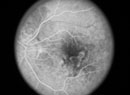
Intraocular lymphoma is a rare entity, and it may masquerade as ocular inflammatory disease, often partially responding to corticosteroid treatment. As a result, diagnosis and treatment can be delayed for years with a significant impact on both vision and mortality. Therefore, awareness of this disease and the subtleties of its presentation, along with clinical suspicion, is critical. In this article, we’ll share diagnostic tips and treatment strategies for this insidious type of cancer.
Definitions and Classifications
Intraocular lymphoma is categorized as vitreoretinal or uveal based on the site involved, with the latter subdivided into choroidal, ciliary body or iris. In addition, it’s classified as either primary, when only the eye is involved, or secondary when it’s associated with pre-existing systemic or central nervous system lymphomas. Primary vitreoretinal lymphoma (PVRL) is closely related to primary central nervous system lymphoma (PCNSL), and is an aggressive malignancy. In contrast, primary uveal lymphomas are more typically indolent, similar to ocular adnexal or orbital lymphomas in terms of aggressiveness.
The predilection of lymphocytes for cancer lies in the essence of their function. Early in the development of both T and B lymphocytes, the genes responsible for antigen recognition undergo rearrangements, insertions and deletions, resulting in an enormous repertoire of immune cells. Upon antigen recognition, B cells undergo rapid proliferation and additional somatic hypermutation of immunoglobulin and non-immunoglobulin genes, as well as double-stranded breaks and recombination during class switching, in order to produce highly specific antibodies. Concurrently, normal cellular checks on mutagenesis, including DNA-damage responses and apoptosis, are downregulated, and cellular differentiation is transcriptionally repressed.
These processes, critical to the immune system’s ability to eliminate pathogens, also place T and B-lymphocytes at risk for the chromosomal translocations and other mutations that characterize lymphomas. These processes typically occur within germinal centers, and can give rise to the likes of diffuse large B cell lymphoma (DLBCL), the subtype that is most commonly found in PVRL. Similar processes occur outside the germinal centers at sites of repeated pathogen exposure, potentially giving rise to marginal zone (MZ) or mucosal-associated lymphoid tissue (MALT) lymphomas. Specifically, repeated antigen stimulation associated with orbital infection by Chlamydia pistaccii or Sjögren’s syndrome can give rise to extranodal marginal zone lymphomas (EZML)1,2 more typical of orbital lymphoma, as well as many uveal lymphomas.
I. PVRL
Following are the salient features of primary vitreoretinal lymphoma:
• Clinical presentation. PVRL presents as vitritis without retinal involvement in middle-aged or elderly patients, and rarely in younger patients. It has no clear racial or gender predilection.3 The presentation and clinical examination findings can vary, but patients most commonly complain of blurred vision and floaters. There is usually moderate vitreous cell, appearing as large homogeneous single cells, and haze, as well as retinal pigment epithelium pigmentary changes, with a quiet anterior segment. There are obvious yellow subretinal infiltrates in half of patients which can sometimes lead to solid RPE detachments. Focal nodular projections from the RPE or sub-RPE infiltrates can be seen on optical coherence tomography in most eyes and fluorescein angiography may reveal hypoflourescent focal lesions. Macular edema and retinal vascular leakage are rare findings, and the optic nerve is variably involved. Although the anterior segment is often quiet, iritis and keratic precipitates can be seen.4 There are myriad atypical findings, including retinal vasculitis, inflammatory glaucoma, neurotrophic keratopathy, choroidal detachment, retinal degeneration, hyphema, hypopyon, retinal vein occlusion and optic disc edema.5
• T cell primary vitreoretinal lymphoma. T cell PVRL presents with anterior uveitis more commonly than B cell PVRL and is more likely to have invaded the iris. CNS involvement occurs at rates similar to B-PVRL, but the presence of systemic lymphoma is more common. The tumors are generally less responsive to methotrexate, though this agent can be used alongside other chemotherapies; it results in less-common remission than in B-PVRL. Mortality is variable, but generally survival is less than one year.6
Adult T cell leukemia/lymphoma (ATL), caused by the human T-lymphotrophic virus type 1 (HTLV-1), is a rare cause of intraocular lymphoma associated with cutaneous and CNS lymphoma. The HTLV-1 virus is endemic in Japan, the Caribbean and Central Africa; it causes anterior, intermediate or panuveitis, as well as a spastic paresis and myopathy. ATL is rare among HTLV-1 carriers, presenting in less than 5 percent of patients in the fourth to sixth decades of life. It’s characterized by deep retinal infiltrates starting in the mid-periphery and progressing posteriorly, which distinguish this entity from HTLV-1 uveitis.7 Vasculitis and symptomatic vitritis, as well as leopard spotting on FA, can be seen. Beyond the retina and vitreous, corneal, conjunctival and orbital tumor involvement have been described.8 ATL presents as one of four subtypes along a transformation continuum, from benign to aggressive, with the latter resistant to chemotherapy and associated with mortality within four to six months.8
Diagnosis
Diagnosis of intraocular lymphoma was historically made by the histologic finding of atypical lymphoid cells with large irregular nuclei and prominent nucleoli isolated from the vitreous.
These studies were augmented with immunohistochemical staining primarily for B and T lymphocyte markers. Modern methodology involves flow cytometry, which allows the simultaneous assessment of multiple surface markers to profile the sample more completely. Additionally, polymerase chain reaction can be used to detect clonal rearrangements in immunoglobulin genes or the T cell receptor. Finally, analysis of extracellular interleukin (IL)-10 and the ratio of IL-10 to IL-6 via aqueous or vitreous tap has been used to indicate lymphoma with sensitivities of 0.80 to 0.90 and specificities of 0.90 to 1.00.4,9 IL-10 is a regulatory cytokine more strongly associated with anti-tumor immune responses, whereas IL-6 is central to most infectious and inflammatory uveitic responses. While the relative elevation in these cytokines is often a good discriminator of neoplastic versus inflammatory processes, it is not an absolute diagnostic indicator, as demonstrated by several researchers, including Esen Akpek, MD, and her group at the Wilmer Eye Institute.10
Undiluted samples as large as 4.5 ml can be obtained using a standard three-port, 20- to 25-gauge vitrectomy with the infusion turned off so as not to dilute the specimen. Air or perfluoron infusion can be used during the procedure to prevent hypotony while maintaining an undiluted specimen. The
 |
diagnostic yield is reported to be between 10 and 75 percent using this technique.11 (This variability is likely a due to variations in specimen processing.) Alternatively, vitreous samples can be obtained in the office via a vitreous tap, or with a 23-ga. handheld portable vitrectomy unit (Intrector, Insight Instruments, Stuart, Florida),12 though this approach is limited by the smaller sample size and hasn’t been validated in cases of suspected intraocular lymphoma.
The challenges to cytologic diagnosis of PVRL from vitreous samples include the large infiltration of reactive T lymphocytes and macrophages, which can outnumber lymphoma cells, as well as the fragility of the lymphoma cells. Intuitively, it might seem that gentle aspiration through a large-bore needle or larger-gauge vitrector may preserve lymphoma cell integrity better than smaller-gauge vitrectomy, but the superiority of one technique hasn’t been conclusively demonstrated.11 Instead, immediate addition of cell culture media supplemented with fetal calf serum, cold transport and rapid processing is recommended to optimize the diagnostic yield.13 Importantly, inclusion of multiple parameters (histology, flow cytometry, cytokine and PCR analysis) will increase the diagnostic yield.
Preoperative consultation with the pathologist is also critical. Ideally, specimens would be obtained before treatment with corticosteroids; but these cases are often only suspected after an inadequate or transient steroid response. Despite these efforts, repeat vitrectomy or retinal/chorioretinal biopsy may ultimately be required.
Approximately 65 to 90 percent of patients with PVRL will have PCNSL at presentation, or will eventually develop it.3 For this reason, thorough evaluation and careful monitoring of patients for CNS involvement is critical. Contrast-enhanced magnetic resonance imaging should be performed as well, and cerebrospinal fluid analysis must be obtained in consultation with an oncologist. CSF cytological analysis, although generally low-yield, may spare the patient a more invasive vitreous or chorioretinal biopsy.
Treatment
PVRLs are generally radiosensitive and respond well to methotrexate14 and, in the case of B cell PVRL, rituximab.15 Treatment is best coordinated with an oncologist and may ultimately reflect patient preferences. Though there have been no randomized controlled treatment trials, the recommendations of the International Primary CNS Lymphoma Collaborative Group Symposium are to treat unilateral ocular disease with local therapy composed of intravitreal methotrexate and rituximab, either alone or in combination with 30 to 35 Gy external beam radiation therapy. Bilateral ocular disease can be treated locally or with systemic chemotherapy, preferentially with adjunctive intravitreal chemotherapy. Finally, when the CNS is involved, systemic high-dose methotrexate and rituximab, along with intravitreal chemotherapy, is recommended, since systemic therapy has limited vitreous penetration. Whole-brain irradiation with ocular radiotherapy is reserved for chemoresistant disease.3
The visual prognosis for PVRL is generally limited unless treatment is started shortly after clinical symptoms begin, and ocular recurrence occurs in 22 percent of cases after treatment.16 CNS disease occurs concurrently or following diagnosis of PVRL in 65 to 90 percent of patients, and is thought to represent multiple, often subclinical, foci of concurrent disease.3 Systemic therapy, therefore, is advocated by some to treat or prevent subclinical disease. Typical treatment for CNS lymphoma is an intense regimen of methotrexate-based intravenous chemotherapy, which is often combined with intrathecal chemotherapy and/or whole-brain radiation and carries significant morbidity. The largest, albeit retrospective, study of PVRL to date16 involved 78 patients and found no significant difference in the occurrence of CNS disease between patients treated locally, systemically or with a combination approach. On average, 36 percent of patients developed CNS involvement in this European study, compared to 47 percent of individuals in an international study,17 which also found no difference in CNS involvement between treatment groups.
II. Uveal Lymphoma
For uveal lymphoma, there are some diagnostic clues and aspects of treatment to keep in mind:
• Clinical presentation. While PVRL has no gender predilection, uveal lymphoma, on the other hand, presents more commonly in men, in the sixth and seventh decades of life, most commonly with blurry vision or no symptoms. The presentation is usually unilateral, though bilateral disease in tertiary referral centers has been reported in up to 54 percent of cases.18 Most patients don’t have systemic lymphoma. Ocular findings include multifocal, creamy, yellow-white choroidal lesions,19 often with early hypofluorescence and late hyperfluorescence on FA; chorioretinal folds, obscuration of choroidal vessels, retinal detachments and, less commonly, uveal effusion.18
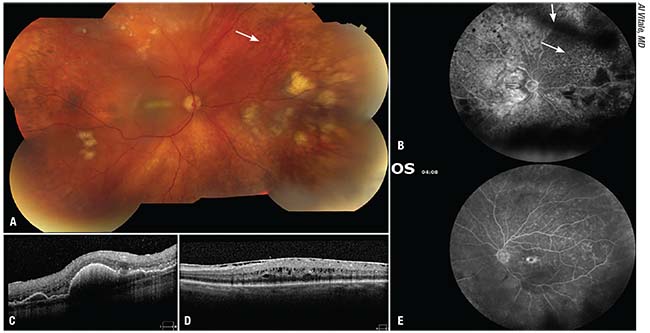 |
| Figure 1. A 70-year-old woman presented with a history of blurry vision and constant sparkly flashes in the right eye which began more than one year prior. She had been treated with cataract surgery, then voriconazole after she developed vitritis and white creamy subretinal lesions. When her lesions failed to respond and she developed vitritis in the left eye, she was referred to our institute. On presentation, her vision was counting fingers in the right eye and 20/50 in the left. Examination revealed mild anterior chamber inflammation with fine keratic precipitates and lens deposits, 1+ vitritis, 2+ haze and inferior snowballs, as well as (A) a macular fold and scattered creamy yellow-white subretinal infiltrates, as well as leopard-spot RPE changes (arrow). (B) Fluorescein angiography of the right eye revealed blocking by the larger subretinal lesions as well as the vitreous debris (arrowhead), mild patchy staining in the macula and patchy window defects in the area of leopard-spot RPE changes (arrow). (C) OCT through one of the nasal lesions revealed punctate hyperreflectivity possibly representing lymphoma cells in the outer retina and pigment epithelial detachments with sub-RPE deposits of hyperreflective material. Examination of the left eye showed 0.5+ vitreous cell and haze, an unremarkable fundus other than the cystoid macular edema, seen also on OCT (D) with punctate hyperreflective foci similar to those seen in the right eye. (E) FA revealed petaloid foveal leakage and diffuse patchy staining throughout the retina. Vitreous and subretinal biopsy from the right eye revealed CD19+CD5+CD10- B cell lymphoma cells. MRI revealed no lesions and the patient was treated with intravitreal methotrexate (400 mcg/0.1 ml) and rituximab (1 mg/0.1 ml). She subsequently developed headache, facial pressure and imbalance, as well as a left visual field defect and was found to have involvement of the posterior right temporal and left frontal lobes. She was then treated with systemic high-dose methotrexate and rituximab. |
The differential diagnosis is broad and includes amelanotic uveal melanoma and metastases, as well as infections, particularly tuberculosis, sarcoidosis, multifocal choroidopathies, posterior scleritis and uveal effusion.
Ocular adnexal involvement or extension is common; it often manifests as a pink subconjunctival or sub-Tenon’s lesion. Additional features, more common in iris and ciliary body lymphoma, include anterior segment inflammation, glaucoma/ocular hypertension caused by infiltration of the angle structures, elevation in episcleral venous pressure by extraocular extension and rubeosis with angle closure.19
OCT reveals retinal changes that reflect the underlying choroidal tumor growth, with smaller tumors associated with a smooth or calm retinal contour and larger tumors associated with a rippled or undulating retinal topography, as well as irregularities in the RPE and outer retina.20 Enhanced-depth-imaging OCT may give details on the underlying tumor. B-scan ultrasound reveals an acoustically hollow choroidal thickening, measuring from 0.9 to 8.9 mm, and variable vitritis.21
Iris lymphomas present with blurred vision and painful red eyes. Examination reveals anterior chamber inflammation or pseudohypopyon, keratic precipitates, posterior synechiae and ciliary injection. While these tumors can masquerade as anterior uveitis, several distinguishing features include infiltrative iris or ciliary body lesions, aberrant iris vessels, hyphema and conjunctival salmon-patch lesions. Lymphoid tumors of the iris can be nonmalignant lymphoproliferative lesions, but they’re often higher-grade B cell or even T cell lymphomas. In one series, about half of the patients had primary iris lymphomas and half had systemic metastases.22
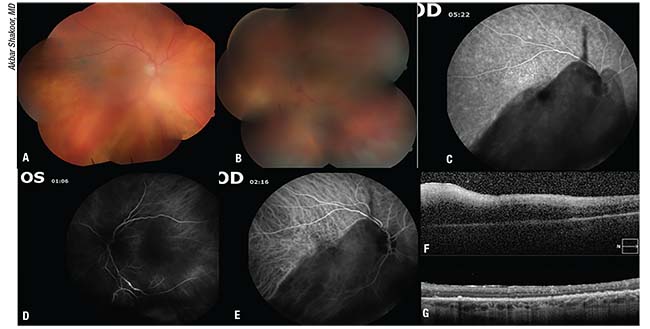 |
| Figure 2. An 89-year-old man presented with a “cobweb” in his left eye for six months that didn’t improve with topical or sub-Tenon’s steroids, as well as new floaters in the right eye. On presentation, his vision was 20/30 in the right eye and hand-motion with an afferent pupillary defect in the left. Examination showed 2+ vitreous cells with mild macular pigment changes in the right eye (A) and 3+ vitreous cells and haze, disc edema/infiltrate, obliterative vasculitis and retinal whitening in the left (B). FA showed mild leopard-spot granularity in the right macula (C) and an occlusive vasculitis in the left eye (D). ICG showed a few hypopigmented spots in the right macula (E). An OCT through the left macula showed a diffuse macular infiltrate (F). The right macula had small subretinal deposits (not shown). Lymphoma was high on the broad differential and the patient was started on oral sulfamethoxazole/trimethoprim and valgancyclovir, and a diagnostic vitrectomy with intravitreal injection of methotrexate, ganciclovir, foscarnet and clindamycin was performed the following day. The vitrectomy specimen revealed only CD19+CD5-CD10- weak kappa large B cells. Workup for systemic or CNS lymphoma was negative. The diffuse retinal infiltrate improved after three weeks (G) although his vision never improved, likely due to the optic nerve involvement. The patient was further treated with intravitreal methotrexate and rituximab in both eyes until he developed an occlusive vasculopathy in the right eye after rituximab injection. He was continued on intravitreal methotrexate for nearly a year before developing a pars plana lesion which is concerning for recurrence in the right eye. |
• Pathophysiology. Debatably regarded as lymphoid hyperplasia, lymphoid neoplasia and extranodal marginal zone lymphoma (EMZL), immunohistochemistry and PCR results suggest that uveal lymphomas represent a spectrum from lympho-proliferative lesions to low-grade lymphomas. They are primarily IgM+, usually monotypic with respect to Ig light chain, with variable levels of similarities to systemic EMZL. Some cases demonstrate monoclonality by PCR. Interestingly, the extraocular extensions are usually composed of more benign-appearing cells or even completely reactive infiltrates23 and the reactive cells are primarily small CD20+ B cells with far fewer reactive T cells than in PVRL.24
• Secondary uveal lymphoma. The most common secondary or metastatic choroidal lymphoma is DLBCL, followed by multiple myeloma, B cell chronic leukemia,
extramedullary plasmacytoma, lymphoplasmocytic lymphoma, EMZL and Waldenstrom’s macroglobulinemia.3,21 Iris and ciliary body involvement is much more common in secondary uveal lymphoma (20 to 30 percent vs. only 4 to 8 percent in primary uveal lymphoma) and these anterior uveal lymphomas tend to be somewhat higher-grade malignancies.21
Diagnosis
Diagnosis of uveal lymphoma can sometimes be made by sampling the epibulbar, conjunctival or orbital components of the disease. However, as noted above, these are more likely to be lymphoproliferative or reactive inflammatory cells than the uveal components.
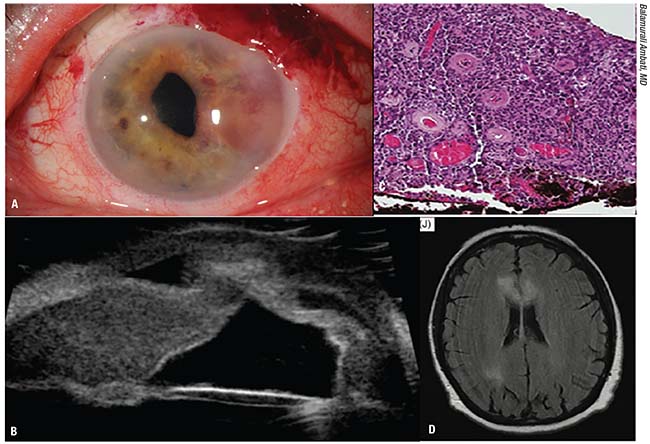 |
| Figure 3. A 77-year-old woman presented with blurry vision, pain and tearing in the left eye. Her visual acuity measured 20/20 in the right eye and 20/60 in the left. Intraocular pressure was 11 mmHg OD and 25 mmHg OS. She had 2+ cell in the left eye with mutton-fat keratic precipitates throughout the inferior cornea. She was treated with topical steroids and cycloplegia for a granulomatous unilateral uveitis. Her eye pressure rose to 52 mmHg and she eventually underwent glaucoma filtration surgery and cataract extraction. When she returned three months later, there was a neovascular infiltrative iris lesion as well as vitritis (A). Ultrasound revealed iridocorneal touch and retro-iridal opacities suggestive of a lymphoproliferative or inflammatory lesion (B). She received an injection of bevacizumab and underwent a surgical iris biopsy which revealed iris stroma diffusely infiltrated by medium-sized lymphocytes with occasional clumped chromatin and rare nucleoli (C). Flow cytometric findings were most consistent with a diffuse large B-cell lymphoma, with high proliferative rate (>90 percent by MIB-1 immunohistochemical stain). The flow cytometric immune profile was positive for CD20, BCL-2, BCL-6 and MUM-1, negative for CD5 and CD10. The proliferative fraction was 90 percent. An MRI revealed right parietal and bilateral corpus callosum lesions and multifocal CNS involvement (D). The patient refused systemic treatment and was treated only locally with methotrexate. She later passed away from complications related to systemic lymphoma. |
Alternatively, samples can be obtained via transvitreal or transscleral needle aspiration of choroidal lesions, or by direct iris or ciliary body biopsy. The vitreous component is also more commonly lymphoproliferative or reactive, and not necessarily diagnostically useful. Systemic investigation for lymphoma should include MRI of the brain as well as complete blood count, serum protein electrophoresis, abdominal and chest computed tomography or PET scan, and bone-marrow biopsy.
Treatment
The prognosis for primary choroidal lymphoma is very good, with the vast majority achieving complete remission after radiotherapy and stability or incomplete remission after chemotherapy. The prognosis for metastatic choroidal lymphoma depends on the primary diagnosis, but a third of patients succumbed to their cancer in one study.21 While eventual CNS involvement is fairly common in PVRL, development of systemic or CNS lymphoma is rare in primary choroidal lymphoma.25,26
In summary, though intraocular lymphomas are rare and clinically variable, certain scenarios should raise suspicion for these potentially devastating tumors. In particular, isolated chronic vitritis, unilateral multifocal choroidal lesions and poorly responsive unilateral posterior uveitis in patients older than 40 should prompt further evaluation, such as ultrasound, FA, OCT of individual lesions and vitreous biopsy, or referral to a uveitis specialist or ocular oncologist. REVIEW
Dr. Hassman is a uveitis fellow and Dr. Shakoor is an assistant professor at the Moran Eye Center at the University of Utah. Neither author has a financial interest in any product mentioned in the article.
1. van den Brand M, Scheijen B, Hess CJ, van Krieken JHJ, Groenen P. Pathways towards indolent B-cell lymphoma - Etiology and therapeutic strategies. Blood Rev 2017;31:6:426-435.
2. Coupland SE. Molecular pathology of lymphoma. Eye (Lond) 2013;27:2:180-189.
3. Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: A report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist 2011;16:11:1589-1599.
4. Davis JL. Intraocular lymphoma: A clinical perspective. Eye (Lond) 2013;27:2:153-162.
5. Abusamra K, Oray M, Ebrahimiadib N, Lee S, Anesi S, Foster CS. Intraocular lymphoma: Descriptive data of 26 patients including clinico-pathologic features, vitreous findings, and treatment outcomes. Ocul Immunol Inflamm 2016:20:1-6.
6. Chaput F, Amer R, Baglivo E, et al. Intraocular T-cell lymphoma: Clinical presentation, diagnosis, treatment, and outcome. Ocul Immunol Inflamm 2017:25:5:639-48.
7. Merle H, Hage R, Meniane JC, et al. Retinal manifestations in adult T-cell leukemia/lymphoma related to infection by the human T-cell lymphotropic virus Type-1. Retina 2016;36:7:1364-1371.
8. Liu MM, Furusato E, Cao X, Shen D, Chan CC. Ocular manifestations and pathology of adult T-cell leukemia/lymphoma associated with human T-lymphotropic virus type 1. Rare Tumors 2010;2:4:e63.
9. Wang Y, Shen D, Wang VM, Sen HN, Chan CC. Molecular biomarkers for the diagnosis of primary vitreoretinal lymphoma. Int J Mol Sci 2011;12:9:5684-5697.
10. Akpek EK, Maca SM, Christen WG, Foster CS. Elevated vitreous interleukin-10 level is not diagnostic of intraocular-central nervous system lymphoma. Ophthalmology 1999;106:12:2291-2295.
11. Kanavi MR, Soheilian M, Hosseini SB, Azari AA. 25-gauge transconjunctival diagnostic vitrectomy in suspected cases of intraocular lymphoma: A case series and review of the literature. Int J Ophthalmol 2014;7:3:577-581.
12. Koch FH, Koss MJ. Microincision vitrectomy procedure using Intrector technology. Arch Ophthalmol 2011;129:12:1599-1604.
13. Ranty ML, Laurent C, Aziza J, et al. Improving the cytological diagnosis of intraocular lymphoma from vitreous fluid. Histopathology 2015;67:1:48-61.
14. Smith JR, Rosenbaum JT, Wilson DJ, et al. Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvement. Ophthalmology 2002;109:9:1709-1716.
15. Larkin KL, Saboo US, Comer GM, et al. Use of intravitreal rituximab for treatment of vitreoretinal lymphoma. Br J Ophthalmol 2014;98:1:99-103.
16. Riemens A, Bromberg J, Touitou V, et al. Treatment strategies in primary vitreoretinal lymphoma: A 17-center European collaborative study. JAMA Ophthalmol 2015;133:2:191-197.
17. Grimm SA, Pulido JS, Jahnke K, et al. Primary intraocular lymphoma: An International Primary Central Nervous System Lymphoma Collaborative Group report. Ann Oncol 2007;18:11:1851-1855.
18. Aronow ME, Portell CA, Sweetenham JW, Singh AD. Uveal lymphoma: Clinical features, diagnostic studies, treatment selection, and outcomes. Ophthalmology 2014;121:1:334-341.
19. Coupland SE, Foss HD, Hummel M, Stein H. Extranodal marginal zone B-cell lymphoma of the lacrimal gland associated with crystal-storing histiocytosis. Ophthalmology 2002;109:1:105-110.
20. Shields CL, Arepalli S, Pellegrini M, Mashayekhi A, Shields JA. Choroidal lymphoma shows calm, rippled, or undulating topography on enhanced depth imaging optical coherence tomography in 14 eyes. Retina 2014;34:7:1347-1353.
21. Mashayekhi A, Shukla SY, Shields JA, Shields CL. Choroidal lymphoma: Clinical features and association with systemic lymphoma. Ophthalmology 2014;121:1:342-351.
22. Mashayekhi A, Shields CL, Shields JA. Iris involvement by lymphoma: A review of 13 cases. Clin Exp Ophthalmol 2013;41:1:19-26.
23. Cockerham GC, Hidayat AA, Bijwaard KE, Sheng ZM. Re-evaluation of “reactive lymphoid hyperplasia of the uvea”: An immunohistochemical and molecular analysis of 10 cases. Ophthalmology 2000;107:1:151-158.
24. Erickson B, Mantopoulos D, Schoenfield L, Cebulla CM. Multimodal imaging and clinicopathologic correlation in primary uveal lymphoma. Case Rep Ophthalmol 2016;7:1:39-43.
25. Dirani A, Allaire G, Callejo S, et al. Choroidal extranodal marginal zone lymphoma diagnosed by full-thickness retinochoroidal biopsy: Case report and review of the literature. Int Med Case Rep J 2017;10:153-158.
26. Bayraktar S, Stefanovic A, Montague N, Davis J, Murray T, Lossos IS. Central nervous system manifestations of marginal zone B-cell lymphoma. Ann Hematol 2010;89:10:1003-1009.