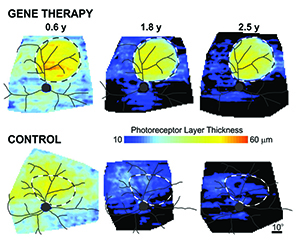
 |
| Using gene therapy to augment normal levels of the RPGR gene resulted in long-term rescue of photoreceptors within the retinal region of gene therapy injection, but not within the control injection. The rescue lasted at least 2.5 years when the disease was treated in its later stages. (Image courtesy University of Pennsylvania.) |
Because the disease affects humans in almost the same fashion as it does dogs, the results suggest that this treatment could be effective and lasting in humans and could set the stage for safety studies that precede a human clinical trial.
“The 2012 study showed that gene therapy was effective if used as a preventive treatment or if you intervene right after the onset of cell death,” said William A. Beltran, DVM, PhD, co-lead author and associate professor of ophthalmology at Penn’s School of Veterinary Medicine. “That was obviously very encouraging. But now we’ve gone further, showing that the treatment is long-lasting and effective even when started at mid- and late-stage disease.”
“This happens to be a very severe disease with very early onset in the first two decades of life in humans,” said Artur V. Cideciyan, PhD, co-lead author and research professor of ophthalmology in the Scheie Eye Institute at Penn’s Perelman School of Medicine. “Because the progression of disease in dogs matches up with the progression in humans, this gives us a lot of confidence about translating these results to eventually treat humans.”
The work involved a close collaboration between Drs. Beltran and Cideciyan as well as Samuel G. Jacobson, professor of ophthalmology at Scheie, and Gustavo D. Aguirre, VMD, PhD, the paper’s senior author and professor of medical genetics and ophthalmology at Penn Vet. The Penn researchers have also long partnered with University of Florida scientists led by William Hauswirth, PhD, the Rybaczki-Bullard Professor of Ophthalmology in the College of Medicine. Their work appears in Proceedings of the National Academy of Sciences.
X-linked retinitis pigmentosa, or XLRP, arises primarily from mutations in the RPGR gene, leading to progressive vision loss starting at a young age. Because it is an X chromosome-linked recessive disease, it overwhelmingly affects boys and men. It is one of the most common forms of inherited retinal disease.
Though rigorously studied, little is understood about the function of RPGR. It is believed to play a role in the function of the connecting cilium, a structure that is present in both rod and cone cells, the photoreceptor cells involved in dim-light and bright-light vision, respectively.
In XLRP, these photoreceptor cells progressively degenerate and die. To counter this effect, the Penn group’s earlier gene therapy work used a viral vector to deliver a normal copy of RPGR specifically to rods and cones using a subretinal injection.
In the new publication, the team reports that the therapy, which occurred when dogs were 5 weeks old, successfully stopped photoreceptor cell loss and maintained vision in dogs for more than three years of study.
This study also went further, using the same viral vector and same approach, except this time beginning the gene therapy intervention at two later time points: At 12 weeks of age, which the researchers term “mid-stage disease,” when approximately 40 percent of the eye’s photoreceptor cells have already died, or at 26 weeks of age, “late-stage disease,” when about 50 to 60 percent of the rods and cones were lost.
The team had concerns about treating at these later stages, both that the retina might not properly reattach following the therapeutic subretinal injection and that there could be toxicity from the viral vector due to the greater extent of photoreceptor cell degeneration. They saw no indications of either being a problem in their follow-up.
“We have spent a lot of time working to make sure the therapeutic gene is tightly regulated in terms of when and where it is expressed,” said Dr. Aguirre. “And, thankfully, we have seen that this therapy appears to be well-tolerated in the retina.”
Instead, what they saw, using non-invasive tests used in human medicine, including electroretinography and optical coherence tomography imaging, was a remarkable and lasting halt in the degeneration of photoreceptor cells in the treated region of the retina. Dogs treated at these later stages of disease even had some of the structural abnormalities in the rods and cones reversed. And these findings translated to improved performance on visual behavior tests, a Y maze that tested whether the dogs could detect a dim light and an obstacle course that assessed their visual navigational skills. The dogs’ performances endured for at least two and a half years after treatment, the latest time point examined, in the late-stage group.
“What the dog studies show, especially those that are treated at a later stage, is that you can treat a relatively small region—20 percent or less of the retinal surface, where you already had 50 percent of photoreceptor cells that died before treatment—and still see not only an electrophysiological improvement and rescue but an actual rescue of visual behavior,” Beltran said.
“Based on my experience developing gene therapies in animal models for many other inherited retinal diseases,” said the University of Florida’s Dr. Hauswirth, “I believe this report describes perhaps the strongest case yet for eventual successful therapy in humans for XLRP.”
As in their earlier work, the researchers showed that the function of both rods and cones was rescued and that these photoreceptor cells were properly connected to the neurons that transmit visual signals to the brain.
“Because this is a photoreceptor disease that affects both rods and cones, or night- and day-vision cells, to show that both were rescued was very wonderful to see,” Dr. Cideciyan said.
“I worry a lot about my patients who have lost photoreceptor cells and possibly have abnormal connectivity and structure in their retina, whether gene therapy would still work for them at later stages of disease,” Dr. Jacobson said. “What we showed here is that the therapy resulted in downstream neurons that were robust and connected, which is exceptionally important for eventual human treatment.”
The group is also studying the other genetic “partners” that function along with RPGR in the connecting cilium to see if there could be additional targets for therapy.
Study Supports Home Monitoring To Detect CNV
Use of a qualification test within a retinal practice appeared to be effective in predicting which patients with intermediate age-related macular degeneration would be good candidates to initiate use of a home monitoring device for progression to more severe AMD, according to a study published online by JAMA Ophthalmology.
Choroidal neovascularization from AMD left untreated or unmanaged after substantial vision loss has occurred remains a leading cause of irreversible blindness in people age 50 years or older throughout much of the world. In the United States, approximately 8 million people have intermediate AMD or monocular advanced AMD, of whom 1.3 million people will develop advanced AMD during the ensuing five years. Patients with intermediate AMD using a home monitoring device (includes looking at a computer screen and using a mouse) have less loss of visual acuity, on average, at detection of choroidal neovascularization than do individuals using standard care monitoring techniques (such as viewing a grid on a piece of paper). Patients must establish a baseline set of responses during a limited series of initial home testing to monitor AMD progression using this device. There is little known about the proportion of patients with high-risk non-neovascular AMD who may be able to incorporate the device successfully into their home monitoring regimen, according to background information in the article.
The developers of the home device designed an in-office qualification test to identify individuals most likely to be able to use the device successfully. Neil M. Bressler, MD, of the Johns Hopkins University School of Medicine, Baltimore, and editor of JAMA Ophthalmology, and colleagues studied 131 participants within a university-based retina practice with intermediate AMD in at least one eye who completed an in-clinic qualification test for the home monitoring device. The qualification test protocol included a short explanation by the study coordinator, explanatory tutorial administered through the device, a trial or practice test administered through the device (an opportunity to mark areas of artificial distortion), an opportunity for the participant to ask questions and the actual qualification test.
A total of 129 participants had reliable qualification test results; 91 participants (70 percent) who completed this test attained a score that suggested they would be able to successfully use the home device. Among the 91 participants who could initiate home testing, 83 did so, including 80 participants (88 percent) who established a baseline value that could be used as a reference for future monitoring. Younger participants were more likely to qualify for home testing. Visual acuity at study enrollment did not appear to be associated with successful qualification.
“These data support the likelihood that a larger percentage of individuals at high risk of progressing to CNV from AMD who successfully complete a qualification test to use this home monitoring device will be able to establish a baseline value for subsequent monitoring at home. These individuals can continue to increase their chance of detecting neovascular AMD between scheduled office visits while the lesion is relatively small and associated with visual acuity that is relatively good,” the authors write.
Sealing Protein May Key Dry-Eye Breakthrough
Recent research by Keck Medicine of the University of Southern California scientists, published in PLOS ONE, suggests a new approach to treating dry eye. Using an experimental mouse model, the researchers demonstrated for the first time that the natural tear protein clusterin seals the ocular surface barrier, while also protecting against further damage.
Findings show that clusterin blocks uptake of fluorescein dye, a clinical test used to diagnose dry eye, according to senior and corresponding author Shinwu Jeong, PhD, assistant professor of research ophthalmology in the Institute for Genetic Medicine at the Keck School of Medicine of USC. “It is well known that clusterin protects cells and proteins,” he said. “A problem in dry eye appears to be that natural clusterin is depleted. We predicted that adding it back would be beneficial, however the novel mechanism of sealing was unexpected.”
The researchers studied the ocular surface barrier rather than upstream effects of tear production, tear chemistry and inflammation that contribute to dry-eye conditions. “We are the first to report functions for this protein in dry eye and shed some light on its potential use for ophthalmology treatments,” said Aditi Bauskar, a PhD, lead author on the paper. “Our pre-clinical results are very promising and make a strong case to use clusterin as a biological drug to prevent or treat not only dry eye, but also other corneal disorders involving damage to the ocular surface barrier.” REVIEW


