The patient was referred with a working diagnosis of idiopathic orbital inflammatory syndrome (IOIS). The differential diagnosis was extensive, including: infectious (e.g., viral, bacterial, mycobacterial and fungal); inflammatory (e.g., sarcoid, IOIS, ruptured dermoid cyst); collagen vascular disease; and benign or malignant tumors. Excisional biopsy was performed. A 20 mm x 18 mm x 6 mm circumscribed firm mass, with a smooth, pink surface, surrounded by fibrosis, was excised. Histopathologic examination revealed a chronically inflamed, scarred and atrophic lacrimal gland with varying degrees of atrophy (See Figure 2). Some acini were well-preserved with others largely replaced by a diffuse infiltrate of well-differentiated lymphocytes and numerous plasma cells. Occasional sections revealed scarce eosinophils. Flow cytometry demonstrated polytypic B cells and CD5 positive T cells with no immunophenotypic evidence of a lymphoproliferative disorder. The patient was diagnosed with chronic inflammation with fibrosis of the right lacrimal gland. Following excisional biopsy, the patient reported resolution of symptoms and did not require systemic treatment.
|
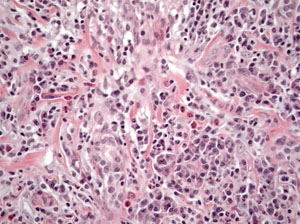
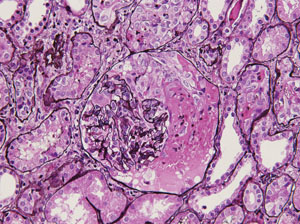
Based on these developments, the differential diagnosis was narrowed to infectious (e.g., tuberculosis) and inflammatory (e.g., Churg-Strauss syndrome, Wegener’s granulomatosis, microscopic polyangiitis, sarcoidosis, and polyarteritis nodosa). Repeat laboratory analysis revealed hemoglobin of 10.8 g/dL, platelets of 456 x 103/μL, ESR of 70 mm/hr, CRP of 2.92 mg/L and complete metabolic panel within normal limits. Additional studies included urinalysis, revealing trace blood, 4+ protein, and trace leukocytes. Testing for anti-nuclear antibody, cytoplasmic-anti-neutrophil cytoplasmic antibody (c-ANCA) and perinuclear-ANCA (p-ANCA), anti-proteinase 3 (anti-PR3), serum angiotensin converting enzyme, tuberculin skin test and group A strep were negative. Anti-myeloperoxidase (anti-MPO) antibody was positive. Based on these results, the patient was diagnosed with Churg-Strauss syndrome. The patient was subsequently treated with cyclophosphamide and prednisone with improvement in her symptoms.
Discussion
Churg-Strauss syndrome is a systemic inflammatory condition traditionally characterized by asthma, eosinophilia and involvement of various organ systems, traditionally the respiratory tract, skin, gastrointestinal tract and kidney.1,2 Churg-Strauss syndrome is classified as an ANCA-associated vasculitis, along with Wegener’s granulomatosis and microscopic polyangiitis.3 While Wegener’s granulomatosis is associated with c-ANCA and anti-PR3 antibodies, Churg-Strauss and microscopic polyangiitis are associated with p-ANCA and anti-MPO antibodies.3,4 Despite these traditional associations, approximately 30 percent of patients with Churg-Strauss express these antibodies.3 Churg-Strauss is also an HLA-associated disease.3 HLA-DRB1*04, HLA-DRB1*07, and HLA-DRB4 confer susceptibility. HLA-DRB1*13 and HLA-DRB3 confer protection.
Ocular findings among the ANCA-associated vasculitides overlap significantly and are not reliably diagnostic of individual diseases. Common symptoms among patients with each condition include: conjunctivitis; episcleritis and scleritis; peripheral ulcerative keratitis; retinal vasculitis; orbital mass, myositis, or dacryoadenitis; nasolacrimal duct obstruction; and neuro-ophthalmic manifestations.3
Treatment of Churg-Strauss, along with other ANCA-associated vasculitides, consists of traditional and novel modalities.1,2 Traditional treatments consist of cyclophosphamide, azathioprine, methotrexate and glucocorticoids. More novel modalities are targeted therapies designed to increased effectiveness while minimizing side effects. These treatments include rituximab, anti-IL-5 antibodies, omalizumab (anti-IgE antibody) and mycophenolate mofetil.1 REVIEW
The author would like to thank Carol L. Shields, MD, Wills Eye Institute Ocular Oncology Service, for her assistance with this case.
1. Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: Update on pathophysiology and treatment. Curr Opin Rheumatol 2012;24:24-30.
2. Abril, A. Churg-strauss syndrome: An update. Curr Rheumatol Rep 2011;13:489-495.
3. Kubal AA, Perez VL. Ocular manifestations of ANCA-associated vasculitides. Rheum Dis Clin N Am 2010;36:573-586.
4. Wilk, A. Clinical and pathophysiological significance of anti-neutrophil cytoplasmic autoantibodies in vasculitis syndromes. Mod Rheumatol 2009;19:590-599.


