Presentation
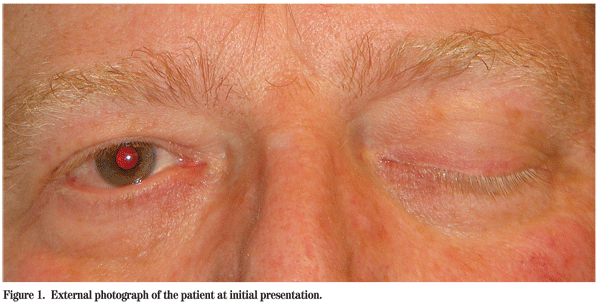
A 53-year-old Caucasian male presented to the Wills Eye Emergency Room complaining of headaches for one week, a two-day history of decreased vision in the left eye and a "droopy" left eyelid (See Figure 1). The patient denied fevers, weight loss, skin infections or recent trauma.
Medical History
The patient had a six-month history of nasal congestion diagnosed as nasal polyposis by an outside otolaryngologist. These symptoms became complicated by two months of recurrent epistaxis requiring nasal packing three days prior to presentation. He also had a history of hypothyroidism, bladder cancer status post resection in 2001, mitral valve prolapse, chronic anxiety and gastroesophageal reflux disease (GERD).
Examination
On examination, the visual acuity in the right eye was 20/25 and 20/200 in the left eye. The patient had complete left-sided ptosis and a left-sided relative afferent pupillary defect. Complete ophthalmoplegia of the left eye was noted with normal extraocular motility of the right eye. Confrontation visual fields were normal in the right eye, but revealed a central scotoma in the left eye. The slit-lamp exam was unremarkable with an intraocular pressure of 18 mmHg in both eyes. The color plate examination was 13/13 in the right eye and 12/13 in the left eye. Hertel exophthalmometry was symmetric in both eyes. The dilated fundus examination was unremarkable.
Diagnosis, Workup and Treatment
The differential diagnosis for a patient with new onset ptosis, decreased vision and complete ophthalmoplegia is extensive. This presentation was worrisome for infiltration of the orbital apex by infectious, inflammatory, traumatic/iatrogenic, vascular or neoplastic etiologies. Of significant concern initially was fungal infection, metastatic disease or possible iatrogenic insults from the patient's recent endonasal procedures. Other possible etiologies included carotid/cavernous sinus disease, sarcoidosis, Wegener granulomatosis, infection or primary neoplasm.
During his early evaluation, this patient received brain and orbital imaging with a noncontrast CT and MRI with and without contrast (See Figure 2). These studies revealed a large sellar and suprasellar mass extending both into the left cavernous sinus and extra-cranially into the nasal cavity. Elevation and stretching of the optic chiasm was also noted along with bony erosion of the sphenoid sinus and clivus. After an unremarkable basic laboratory evaluation, the patient was admitted to neurosurgery with continuing evaluation by neuro-ophthalmology, ENT and endocrinology. Additional studies were ordered to evaluate pituitary function. The prolactin level was elevated at 1,134 ng/mL (normal 0 to 19 ng/mL).
With this new information, the diagnosis of a giant prolactinoma was made and the patient was started on oral cabergoline (a dopaminergic suppressor of prolactin secretion.) Over the next week the patient achieved partial resolution of ptosis, his motility improved, and his visual acuity was 20/30 OS. Within two days, a second MRI (not shown) revealed a measurable decrease in tumor volume and his prolactin decreased to 172 ng/mL.
He was discharged to follow-up with neuro-ophthalmology and endocrinology. His prolactin level continued to decline to 67 ng/mL one week post-discharge. At a six-week follow-up the patient had normal perimetry testing, his visual acuity was stable at 20/30, his ptosis had resolved, and he had only mild residual extra-ocular motility deficits.
Discussion
The prolactinoma is the most common pituitary adenoma and accounts for 30 percent of all pituitary tumors. Those lesions greater than 1 cm in diameter are designated as a macroprolactinoma, and those greater than 4 cm are called giant prolactinomas. This latter type, found in our patient, is quite rare (0.5 percent of all pituitary tumors) and often accompanied by prolactin elevations greater than 1,000 ng/mL and mass effect in the form of compressive optic neuropathy, visual field deficits and ophthalmoplegia. Clinical manifestations are quite variable and depend on sites of invasion. Interestingly, while our patient presented with signs of optic neuropathy and ophthalmoplegia, his visual field defect was central and not the classic bitemporal deficit. The true visual field defect in each patient is sometimes variable and dependent on the relationship between the tumor and the chiasm, pre-chiasmatic optic nerves and post-chiasmatic optic tracts.
Additionally, a major concern with giant prolactinomas (which can quickly increase optic nerve compression and visual field loss) is the development of apoplexy in which central tumor necrosis leads to hemorrhage and rapidly increased mass effect.

Treatment of giant prolactinomas in the past has posed a dilemma for many physicians when attempting to choose between medical and surgical modalities. This situation was further complicated by the rarity of the disease, and thus few studies are available evaluating treatment options. Recent studies have shown the efficacy of dopamine agonist (i.e. bromocriptine and cabergoline) medication as primary treatment for giant prolactinomas. In one large scale retrospective case study, patients all showed visual field improvement with a mean decreased tumor volume of 68 percent. This data supports the contention that most tumors can be effectively managed without surgical decompression given the exquisite sensitivity of prolactinomas to dopamine analogs.
However, surgical decompression is considered standard in cases of apoplexy, rapidly changing visual symptoms and poor tolerance of, or initial response to, dopamine agonist therapy. It has been suggested in the literature that surgical decompression be conducted at or before six weeks status post-initiation of medical therapy given the diminishing chances for success with medication alone at this time point.
Patient follow-up should be close, with evaluation of symptom progression, perimetry, visual acuity and motility. Patients should be followed by an endocrinologist for serial prolactin monitoring and brain imaging. Dopamine agonist therapy is typically a long-term commitment with the possibility of mass recurrence if discontinued.
The author thanks Jennifer Hall, MD, of the Wills Eye Institute Neuro-Ophthalmology Service, for her time and assistance with this case.
1. Wu Z, Yu C, Su Z, Zhuge Q, Wu J, Zheng W. Bromocriptine treatment of invasive giant prolactinomas involving the cavernous sinus: Results of a long-term follow-up. J Neurosurg 2006;104:54-61.
2. Shrivastave R, Aringteanu M, King W, Post K. Giant prolactinomas: Clinical management and long-term follow-up. J Neurosurg 2002;97:299-306.
3. Brisman M, Katz G, Post K. Symptoms of pituitary apoplexy rapidly reversed with bromocriptine, Case Report. J Neurosurg 1996;85(6):1153-5.
4. Liu J, Couldwell W. Contemporary management of prolactinomas. Neurosurg Focus 2004 Apr 15;16(4):E2.
5. Walsh and Hoyt's Clinical Neuro-Ophthalmology, 6th Edition, vol 2; 2005:1533-42.
6. Famularo G, Pozzessere C, Piazza G, De Simone C. Abrupt-onset oculomotor paralysis: An endocrine emeregency. Eur J Emerg Med. 2001;8(3):233-6.
7. Yeh S, Foroozan R. Orbital apex syndrome. Curr Opin Ophthalmol 2004;15:490-498.


