 |
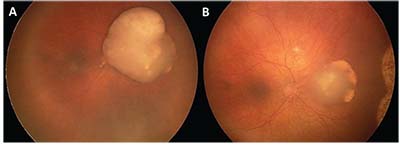
| Figure 1. (A) Nasal retinoblastoma with overlying vitreous seeds treated with three cycles of intraarterial melphalan, showing good tumor response and calcified vitreous seeds (B). |
Retinoblastoma
Retinoblastoma is the most common primary intraocular tumor in children, with an incidence of 11.8 per million in the United States.1 It’s caused by mutations in the tumor suppressor RB1 gene (located on Chromosome 13q14); both alleles
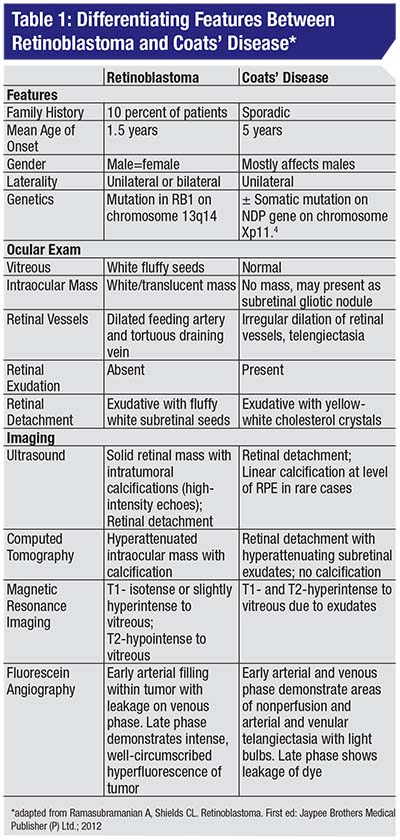 |
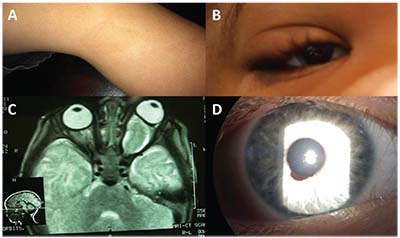
• Clinical diagnosis. The most common presenting sign of retinoblastoma is leukocoria followed by strabismus. On exam the tumor appears as a translucent or white mass, and may be endophytic (into the vitreous cavity), exophytic (towards the subretinal space), or diffusely infiltrating. Ultrasound will show calcification in 90 percent of cases, while CT-scan should be avoided in suspected cases. The differential diagnosis for retinoblastoma includes persistent fetal vasculature, toxocariasis and Coats’ disease (Table 1) (Figure 2A&B).
• Treatment. Patients with unilateral disease may undergo focally destructive therapy (cryopexy, laser photocoagulation, hyperthermia and plaque irradiation) in conjunction with systemic chemoreduction. Recently, intra-arterial chemotherapy is being employed for treatment (Figure 1), while enucleation is reserved for large tumors or recalcitrant cases. Bilateral cases will require systemic chemotherapy.2
• Family screening and genetic counseling. In patients with bilateral retinoblastoma there is a 50-percent chance of the offspring being affected. If these patients have an identified germline mutation, DNA testing can be performed in pregnancy or in neonates to establish the risk to the offspring. If testing of the patient fails to identify a germline mutation, the child must undergo surveillance eye exams.
In patients with unilateral retinoblastoma, a germline mutation may occur in 10 to 15 percent of patients, the risk to an offspring is similarly 50 percent. In the remaining patients, if tumor cells test positive for a somatic mutation, there is less than a 5-percent risk for the affected offspring. In either case, DNA testing can be pursued in pregnancy or in neonates to establish risk. If patients with unilateral retinoblastoma have no germline mutation and no tumor is available for testing, the risk to the offspring is less than 5 percent, no testing is possible and the child must undergo surveillance eye exams.
The recommended screening for familial retinoblastoma is for an initial exam within two weeks of birth, monthly exams up to age 3 months, bi-monthly exams till age 1, tri-monthly exams till age 2,
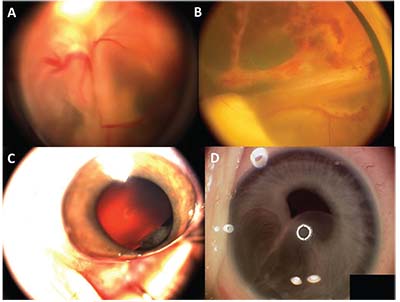 |
| Figure 2. (A) Retinoblastoma with total retinal detachment showing the dilated tortuous vessels that dip in to feed the tumor. (B) Coats’ disease with total retinal detachment showing the telangiectatic vessels in the periphery. (C) Fleshy tumor arising from the ciliary body in a child suggestive of medulloepithelioma. (D) Iris stromal cyst in a child. |
If a germline mutation is identified in a child and the parents tested negative, it’s possible that one parent could have germline mosaicism, in which case testing of siblings is recommended. If mosaicism for germline mutation is identified in the child, the mutation was a post-zygotic event and no testing of siblings is warranted.2
Medulloepithelioma (Diktyoma)
This is a rare embryonic tumor of the non-pigmented ciliary epithelium. It’s the second most common pediatric primary intraocular tumor. The difficult direct visualization of these tumors may lead to late diagnosis, misdiagnosis and improper initial management. Even after symptoms develop, the possibility of a tumor is often overlooked, as patients are treated for secondary complications of the tumor such as cataract or glaucoma. Later, as the tumor slowly grows, it may be seen in the pupil, distort the iris, or invade adjacent tissues. The tumor appears as an irregularly shaped, smooth, fleshy pink lesion arising from the ciliary body (Figure 2C), oftenwith large cystic spaces that contain vitreous-like material. These cysts may break and float off into the aqueous or vitreous humor. Ultrasound biomicroscopy and AS-OCT are useful for visualization of the medium-to-highly reflective ciliary body mass with intratumoral cystic spaces.4 A characteristic retrolental neoplastic cyclitic membrane may be found in half of the cases.5 Histologically it’s classified as non-teratoid or teratoid (it often has hyaline cartilage), and as benign or malignant. Unlike medulloepithelioma of the central nervous system, metastasis is rare. Treatment options include cryotherapy, local resection, plaque radiotherapy and external beam radiotherapy but, often, enucleation is required.
 |
Iris Stromal Cysts
In children, these are often primary cysts; acquired cysts mostly occur in adults.6 In contrast to iris pigment epithelium cysts, these tend to enlarge aggressively, especially in children. Iris stromal cysts appear as a translucent mass occupying the anterior iris, expanding to fill much of the anterior chamber and covering the pupil, leading to angle occlusion and glaucoma (Figure 2D). Occasionally, the cyst can rupture, causing anterior uveitis, secondary glaucoma and an increased risk for epithelial downgrowth. These cysts are often difficult to manage and a variety of treatment strategies have been used, including aspiration, cryotherapy, laser and surgical removal.7,8 Recently, physicians have had considerable success treating these cases with sclerosing therapy using absolute alcohol.9
 |
Phakomatoses
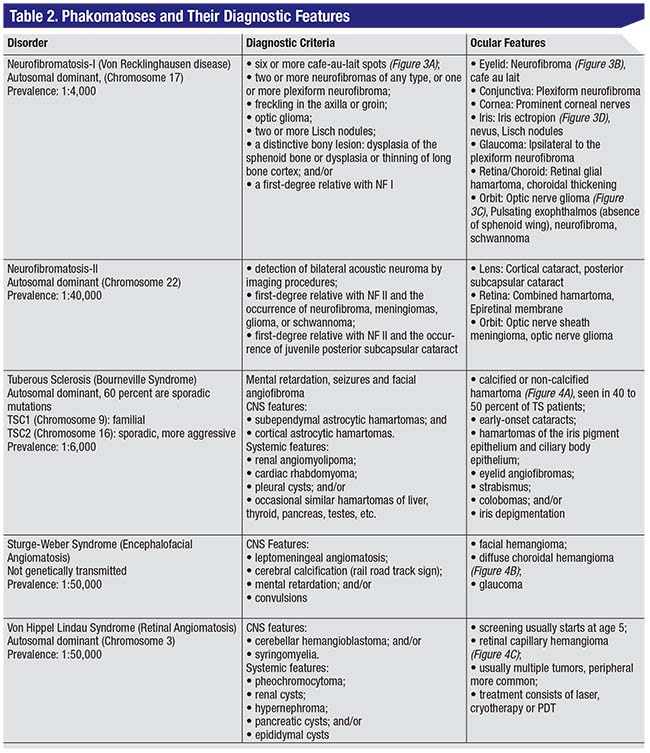
Tumors often occur in combination with neurocutaneous disorders or phakomatoses. Some salient features of common phakomatoses are outlined in Table 2.
In cases of optic nerve glioma (as can occur with neurofibromatosis, seen in Table 2), there are some screening guidelines to note: Although there is some non-uniformity regarding screening frequency and duration in the literature, in most centers screening is performed every six months until age 6 and then annually up to ages 16 to 18.10 This includes visual acuity testing, pupillary reflex testing and fundoscopy, while a brain MRI is usually reserved for when there is clinical suspicion.
 |
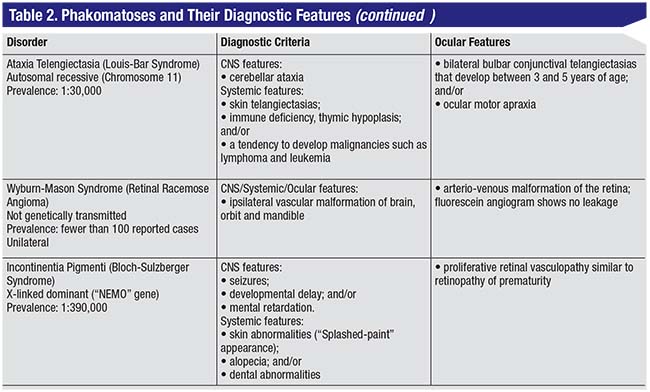
| Figure 3. Features of neurofibromatosis-1 include café-au-lait spots (A), upper eyelid plexiform neurofibroma (B), optic nerve glioma (C) and congenital iris ectropion (D). |
Capillary Hemangioma
These hamartomatous growths composed of proliferating capillary endothelial cells are the most common benign tumors of infancy. They occur in 1 to 3 percent of term newborns, are more common in premature infants and occur in varying frequency across races (10 to 12 percent in white, non-Hispanic infants; 1.4 percent in black infants and 0.8 percent in Asian infants). The salient features are the following:
• Clinical presentation. Most hemangiomas are clinically insignificant at birth. They present as erythematous macules or telangiectasia (Figure 4D). The natural history is of rapid proliferation in the first several months of life, typically followed by regression after a year. Rarely, they may ulcerate or bleed. A large, facial, plaque-like capillary hemangioma involving one or more dermatomes may be associated with an X-linked dominant systemic condition called PHACES Syndrome (posterior fossa brain malformation, facial hemangiomas (segmental, >5 cm), arterial anomalies, cardiac anomalies and aortic coarctation, eye abnormalities, sternal cleft and supraumbilical raphe syndrome).
• Treatment. Indications for intervention include occlusion of the visual axis, induction of astigmatism, corneal exposure due to severe proptosis and optic nerve compression by a rapidly growing tumor. A deep hematoma causing proptosis merits imaging to assess the tumor size. Non-ocular indications include hypertrophy of epidermal and subcutaneous tissue leading to maceration, erosion of epidermis; or even cardiac, hematologic and pulmonary complications, as in the case of a sizeable hematoma. Therapies include beta-blockers (topical or systemic), steroids (intralesional or systemic), surgery, laser and radiation.
We prefer using systemic propranolol as follows: After performing a complete ocular exam and ruling out the possibility of PHACES, the patient is sent to a pediatrician where heart rate, blood pressure, blood sugars and an ECG are performed. If the ECG i
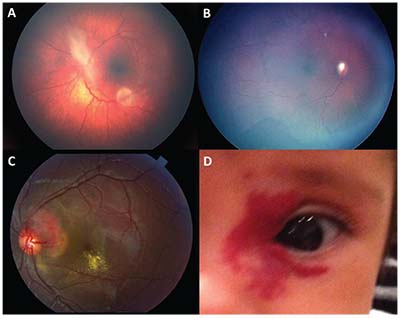 |
| Figure 4. Astrocytic hamartoma in a patient with tuberous sclerosis (A). Diffuse choroidal hemangioma in a patient with Sturge-Weber syndrome, note the dark pigment, optic nerve cupping and peripheral vessel shunting (B). Retinal capillary hemangioma in Von Hippel-Lindau disease (C) and capillary hemangioma (D). |
Side effects of this treatment include bradycardia, hypotension, hypoglycemia and gastrointestinal disturbances. Propranalol should be administered along with feeding and should be withheld if a child is not eating or is vomiting. Therapy is continued until the complete regression of hemangioma occurs or until 9 months of age. The child is then weaned off of this over two to three weeks (similar to initiation) and the hemangioma observed for any signs of recurrence. If there is any recurrence, propranalol is restarted.
Pulsed dye laser may be used to treat residual telangiectasias, and residual texture irregularities can be improved by resurfacing with carbon dioxide or erbium lasers. Topical timolol may be used for superficial hemangiomas. Steroids are rarely used due to the side effects and the efficacy of propranolol. Interferon alpha-2a and/or vincristine may be used in severe cases, and surgery may be required either as a primary or reconstructive procedure. REVIEW
Dr. Sophie is a resident physician and Dr. Ramasubramanian is an assistant professor at the University of Louisville’s Department of Ophthalmology. They have no financial interest in any products mentioned here.
This work was supported in part by an unrestricted institutional grant from Research to Prevent Blindness, New York, N.Y.
1. Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 2009;93:21-3.
2. Ramasubramanian A, Shields CL. Retinoblastoma. First ed. Jaypee Brothers Medical Publisher Ltd.; 2012.
3. Moll AC, Imhof SM, Meeteren AY, Boers M. At what age could screening for familial retinoblastoma be stopped? A register-based study 1945-98. Br J Ophthalmol 2000;84:1170-2.
4. Gunduz K, Hosal BM, Zilelioglu G, Gunalp I. The use of ultrasound biomicroscopy in the evaluation of anterior segment tumors and simulating conditions. Ophthalmologica 2007;221:305-12.
5. Kaliki S, Shields CL, Eagle RC, Jr., et al. Ciliary body medulloepithelioma: Analysis of 41 cases. Ophthalmology 2013;120:2552-9.
6. Shields CL, Kancherla S, Patel J, et al. Clinical survey of 3680 iris tumors based on patient age at presentation. Ophthalmology 2012;119:407-14.
7. Shields JA, Shields CL, Lois N, Mercado G. Iris cysts in children: Classification, incidence, and management. The 1998 Torrence A Makley Jr Lecture. Br J Ophthalmol 1999;83:334-8.
8. Lois N, Shields CL, Shields JA, Mercado G, De Potter P. Primary iris stromal cysts. A report of 17 cases. Ophthalmology 1998;105:1317-22.
9. Shields CL, Arepalli S, Lally EB, Lally SE, Shields JA. Iris stromal cyst management with absolute alcohol-induced sclerosis in 16 patients. JAMA Ophthalmol 2014;132:703-8.
10. Caen S, Cassiman C, Legius E, et al. Comparative study of the ophthalmological examinations in neurofibromatosis type 1. Eur J Paediatr Neurol 2015;19:415-22.


