 |
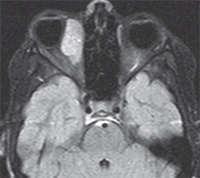
| Figure 2. T1 MRI axial image post contrast demonstrating a right medial mildly lobulated orbital mass measuring 2.8 x 1.7 x 3.1 cm with diffuse enhancement post contrast. |
The differential diagnosis included both inflammatory/infectious lesions as well as malignant and benign mass lesions. The painless nature of her presentation made inflammatory lesions less likely, and the presence of a focal mass decreased the suspicion for infection. Given the rapidly progressive course of the child’s orbital process, magnetic resonance imaging of the brain and orbits with intravenous contrast was obtained on an urgent basis to further characterize the lesion. Imaging revealed a right orbital mass located medially that showed diffuse enhancement with gadolinium (See Figure 2).
Because of the high clinical suspicion for rhabdomyosarcoma and consistent appearance on imaging, the patient underwent urgent biopsy of the lesion. Incisional biopsy with debunking rather than excisional biopsy was pursued given the lesion’s close proximity to the globe and optic nerve posteriorly. The biopsy revealed embryonal cell rhabdomyosarcoma, confirmed with immunohistochemistry (See Figure 3).
The patient was diagnosed with rhabdomyosarcoma and referred to an oncologist for further staging and treatment. Plans were made for the patient to undergo four courses of combination chemotherapy with vincristine, actinomycin and cyclophosphamide followed by weekly treatments of vincristine alone to complete a six-month course. Additionally, she would undergo six weeks of proton beam radiation therapy for local control. On her first postoperative visit, following a single course of chemotherapy treatment, the right orbital mass was no longer clinically detectable. At six-month follow-up she had mild dry-eye syndrome, but was completely tumor-free.
Discussion
Rhabdomyosarcoma arises from pleuripotent mesenchymal cells. It accounts for 5 percent of all childhood cancers and 20 percent of all malignant soft tissue tumors.1 Ocular rhabdomyosarcoma typically arises from the orbital soft tissues and can manifest with a variety of clinical features including proptosis; globe displacement; blepharoptosis; conjunctival and eyelid swelling; a palpable mass; as well as with pain.1 Orbital rhabdomyosarcoma accounts for nearly 25 to 35 percent of head and neck rhabdomyosarcomas.1
This case demonstrates the diagnostic dilemma that may be present in cases of orbital rhabdomyosarcoma, because rhabdomyosarcoma may masquerade as a more benign process before the patient begins to manifest orbital signs. It is important to always consider rhabdomyosarcoma in the scenario of a rapidly growing orbital lesion in children and teenagers. Infections can also progress rapidly, but typically cause generalized inflammation rather than a focal mass lesion, with the exception of a concomitant abscess. Making the diagnosis depends primarily on imaging.1 Therefore, clinical suspicion must remain high so that imaging is pursued in an expedited manner. In a child, care should be taken to limit radiation exposure, making MRI a more attractive modality. However, availability of MRI can be limited, in which case a computed tomography scan may be preferable. The appearance of rhabdomyosarcoma on MRI is certainly not diagnostic but typically manifests as a well-circumscribed homogenous round mass that is isointense to the extraocular muscles on T1 and hyper-intense on T2.1 The most common orbital location is superonasal, but as seen in this case, it may be anywhere.1 Diffusion-weighted images may be helpful in distinguishing malignant orbital tumors from benign lesions.2 The mean apparent diffusion coefficient has been shown to be lower in malignant masses.2 This property is thought to relate to the reduced extracellular matrix and diffusion space in malignant tumors due to their enlarged nuclei and hypercellularity relative to benign tissues.2
In order to confirm the diagnosis, a biopsy must be performed. There has been some debate in the literature regarding the surgical approach in patients with orbital rhabdomyosarcoma.1 While some believe that an attempt should be made to remove the mass in its entirety,1 often there is the consideration of the surrounding critical structures that can make complete excision challenging. Malignant cells lack cohesiveness and are easily detached and freed to start a new malignant colony in normal tissue if the primary tumor is disrupted.3 This property has been demonstrated in the literature, with tumor seeding in breast malignancy following fine- needle aspiration biopsy.3 Increased risk of seeding has likewise been seen when cutting into a mass for incisional biopsy.3 Therefore, for malignancies in general, excisional biopsy is preferred when the tumor can be removed in total with wide margins.3 However, in children with rhabdomyosarcoma, total resection should not be pursued if there is a high risk of functional or cosmetic consequence.4
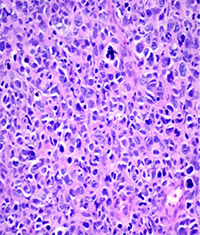 | 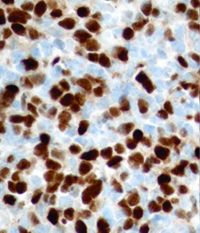 | 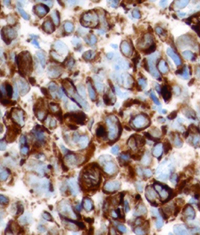 |
| Figure 3. Right orbital mass biopsy. Histopathology section demonstrating embryonal cell rhabdomyosarcoma on hematoxylin and eosin staining (left). Immunohistochemistry demonstrating positivity with antibodies to Desmin (center) and Myogenin (right). | ||
Fortunately, the outcome of patients with orbital rhabdomyosarcoma is quite good.6 The staging of rhabdomyosarcoma is determined by location in addition to tumor size, nodal involvement and metastases. Tumors present in the orbit and elsewhere in the head and neck are all classified as stage one regardless of tumor size or node status. Pathologic subtype of these tumors is another important consideration for patient risk stratification.6 In children, the embryonal and alveolar subtypes are most common. The alveolar subtype is fortunately relatively rare, as it portends a worse prognosis, and these patients are automatically stratified as intermediate or high-risk.6 Distinguishing the morphologic features on histology to determine the rhabdomyosarcoma subtype is not without its challenges. In fact, having any amount of alveolar morphology present in a specimen previously characterized the tumor as alveolar even if it was not the predominant finding.7
Tumor genetics is now reshaping the way rhabdomyosarcoma is histologically classified. Testing for the PAX/FOXO1 fusion gene by molecular methods has become increasingly important for determining prognosis.7 The presence of this fusion gene in patients with the alveolar subtype of rhabdomyosarcoma imparts a worse prognosis, while gene-negative patients have clinical outcomes similar to patients with the embryonal subtype of rhabdomyosarcoma.7 In a retrospective study of 210 cases, fusion gene-negative alveolar cell rhabdomyosarcoma had a similar period of tumor-free survival and frequency of metastasis when compared to embryonal cell rhabdomyosarcoma, whereas patients with fusion gene positive alveolar cell rhabdomyosarcoma had statistically significantly worse outcomes.7
This case highlights the importance of considering rhabdomyosarcoma in young teens; although more common among young children, it can still manifest in older age groups. It is vital to check for orbital signs when these patients present, because as seen in this case, the early manifestations of rhabdomyosarcoma may masquerade as more benign entities. In a disease process where tumor size impacts prognosis,6 and growth in locations such as the orbit may jeopardize the surrounding critical structures, it is vital to expedite diagnosis and intervention to maximize functional outcomes and survival. REVIEW
The author would like to give special thanks and acknowledgement to Robert B. Penne, MD, Richard Schnall, MD, and Ralph C. Eagle Jr., MD.
1. Shields JA, Shields CL. Rhabdomyosarcoma: Review for the ophthalmologist. Surv Ophthalmol 2003;48:39-57.
2. Razek AK, Elkhamary S, Mousa A. Differentiation between benign and malignant orbital tumors at 3-T diffusion MR-imaging. Neuroradiology 2011;53:517-522.
3. Shyamala K, Girish HC, Murgod S. Risk of tumor cell seeding through biopsy and aspiration cytology. J Int Soc Prev Commmunit Dent 2014;4(1):5-11.
4. Stevens M. Treatment for childhood rhabdomyosarcoma: The cost of cure. Lancet Oncol 2005;6:77-84.
5. Ladra M, Edgington S, Mahajan A, Grosshans D, et al. A dosimetric comparison of proton and intensity modulated radiation therapy in pediatric rhabdomyosarcoma patients enrolled on a prospective phase II proton study. Radiother Oncol 2014;113:77-83.
6. Malempati S, Hawkins D. Rhabdomyosarcoma: A review of children’s oncology group (COG) soft tissue sarcoma committee experience and rational for current COG studies. Pediatr Blood Cancer 2012;59:5-10.
7. Williamson D, Missiaglia E, de Reyniès A, Pierron G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 2010;1;28(13):2151-8.


