 | 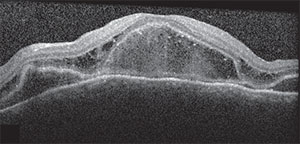 |
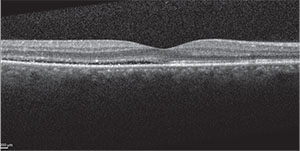
| Figure 2A. OCT of the right macula showing serous detachment temporal to and involving the fovea. | Figure 2B. OCT of the left macula showing RPE undulation, inflammatory membranes and ellipsoid zone disruption. |
Spectral-domain optical coherence tomography demonstrated a small amount of subretinal fluid in the right eye and extensive subretinal fluid with RPE undulation, inflammatory membranes and ellipsoid zone disruption in the left eye (See Figure 2). Fluorescein angiography demonstrated early patchy choroidal filling, pinpoint areas of hyperfluorescence in the posterior pole, pooling of fluorescein in areas of detachment in the left eye and disc hyperfluorescence in both eyes (See Figure 3).
These findings, in addition to the patient’s systemic complaints, were suggestive of the acute uveitic stage of Vogt-Koyanagi-Harada (VKH) disease (also called VKH syndrome). Therapy was initiated with 60 mg of prednisone a day. Ten days after initiating therapy, macular OCT (See Figure 4) showed resolution of subretinal fluid with improved foveal contour in both eyes. The ellipsoid zone continued to show breakage and disappearance, and the patient’s visual acuity in both eyes remained unchanged at this early follow-up.
Discussion
VKH disease is a granulomatous, multisystem, autoimmune disorder affecting tissues with large amounts of melanin pigment, including the eyes, meninges, skin and auditory system. It most commonly affects more heavily pigmented individuals, including Hispanics, Asians, Native Americans, Middle Easterners and Asian Indians. The exact mechanism and sensitizing factor in patients with VKH disease is unknown, although evidence suggests a T-lymphocyte-mediated process against an unidentified antigen of melanocytic cells.1-4
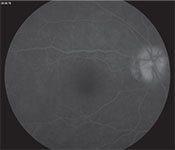 |
| Figure 3A. Late-phase fluorescein angiography of the right eye showing optic disc hyperfluorescence. |
In 2001, an international consensus panel published a revised list of diagnostic criteria for designating VKH disease as “complete” or “incomplete” based on the spectrum of manifestations seen at the time of presentation.1 Ophthalmic criteria include bilateral ocular involvement with no history of ocular trauma or surgery and no evidence suggestive of other ocular disease entities. Systemic manifestations are divided into neurologic/auditory findings including meningismus, tinnitus or cerebrospinal fluid pleocytosis and integumentary findings including alopecia, poliosis or vitiligo. “Complete” VKH disease requires all of the above criteria to be met at some point in the patient’s disease course, while “incomplete” VKH disease requires all ophthalmic criteria as well as either neurologic/auditory or integumentary findings. The panel also allowed for a designation of “probable” VKH disease in patients who met the above ocular criteria in the absence of systemic signs or symptoms.1
The clinical course of VKH disease typically follows four stages. The prodromal stage mimics viral illness and can include headaches, tinnitus, neck stiffness, nausea and hearing loss.3,4 This is followed three to five days later by the acute uveitic stage that can last for several weeks and includes signs of panuveitis, optic disc swelling and choroiditis with exudative retinal detachments. The chronic convalescent stage gradually develops and manifests as depigmentation of the skin and uvea. During the convalescent stage, there may be recurrent episodes of uveitis known as the chronic recurrent stage that lead to vision-threatening complications including cataracts, glaucoma, choroidal neovascular membranes and subretinal fibrosis.3,4
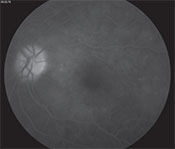 |
| Figure 3B. Late-phase FA of the left eye showing optic disc hyperfluorescence, scattered pinpoint hyperfluorescence and fluorescein pooling in areas of exudative retinal detachment. |
Ophthalmic imaging can be utilized to further characterize clinical findings. Fluorescein angiography most frequently features early patchy choroidal filling, early pinpoint hyperfluorescence in the posterior pole, disc hyperfluorescence and subretinal pooling of fluorescein in areas of exudative retinal detachment.5 Features on OCT that distinguish VKH from other causes of serous retinal detachment include folds of RPE, fluctuation of the internal limiting membrane and subretinal septa disrupting the photoreceptor layer.6 In chronic forms of VKH, the RPE can show signs of thickening and breakage, as well as disappearance of the cone outer segment tip (COST) line or ellipsoid zone.7 The use of OCT with enhanced-depth imaging can provide better visualization of the choroid, which can be thickened in acute-phase and chronic-recurrent phase VKH patients.8,9
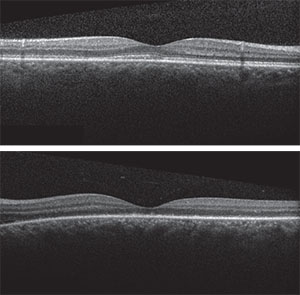 |
| Figure 4A & B. OCTs of the right and left macula showing resolved serous detachment with damage of the ellipsoid zone in the left macula. |
In conclusion, VKH is a multi-system inflammatory disorder with bilateral ocular involvement most commonly manifesting as multifocal exudative retinal detachments due to diffuse choroiditis. Depending on the timing of presentation, there are a variety of neurologic, auditory and integumentary manifestations. The syndrome progresses through numerous stages, and in the acute phase it is most commonly treated with high-dose corticosteroids. Later, a chronic recurrent stage can develop that may require treatment with long-term immunomodulatory agents. The diagnosis is one of exclusion after an appropriate clinical and laboratory evaluation for other uveitic processes that could mimic the inflammatory ocular findings. Due to the systemic nature of VKH, the presence of bilateral panuveitis and features of choroiditis should prompt a detailed review of systems to avoid a delay in treatment. REVIEW
1. Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001;131:647-652.
2. Norose K, Yano A. Melanoma specific Th1 cytotoxic T lymphocyte lines in Vogt-Koyanagi-Harada disease. Br J Ophthalmol 1996;80(11):1002-1008.
3. Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol 1995;39(4):265-292.
4. Burkholder BM. Vogt-Koyanagi-Harada disease. Curr Opin Ophthalmol 2015;26(6):506-511.
5. Chee SP, Jap A, Cheung CM. The prognostic value of angiography in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2010;150:888-893.
6. Lin D, Chen W, Zhang G, et al. Comparison of the optical coherence tomographic characters between acute Vogt-Koyanagi-Harada disease and acute central serous chorioretinopathy. BMC Ophthalmol 2014;14:87-2415-14-87.
7. Zhou M, Jiang C, Gu R, Sun Z, Huynh N, Chang Q. Correlation between retinal changes and visual function in late-stage Vogt-Koyanagi-Harada disease: An optical coherence tomography study. J Ophthalmol 2015;2015:916485.
8. Tagawa Y, Namba K, Mizuuchi K, et al. Choroidal thickening prior to anterior recurrence in patients with Vogt-Koyanagi-Harada disease. Br J Ophthalmol 2016;100(4):473-477.
9. Fong AH, Li KK, Wong D. Choroidal evaluation using enhanced depth imaging spectral-domain optical coherence tomography in Vogt-Koyanagi-Harada disease. Retina 2011;31:502-509.
10. Read RW, Yu F, Accorinti M, et al. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2006;142:119-124.
11. Jabs DA, Rosenbaum JT, Foster CS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: Recommendations of an expert panel. Am J Ophthalmol 2000;130:492-513.
12. Lai TY, Chan RP, Chan CK, Lam DS. Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada disease. Eye (Lond). 2009;23(3):543-548.
13. Abu El-Asrar AM, Hemachandran S, Al-Mezaine HS, et al. The outcomes of mycophenolate mofetil therapy combined with systemic corticosteroids in acute uveitis associated with Vogt-Koyanagi-Harada disease. Acta Ophthalmol 2012;90(8):e603-8.


