Workup, Diagnosis and Treatment
 |
Given the radiographic appearance and painless, chronic, slow-growing nature of the lesion, the most likely diagnosis was felt to be orbital cavernous hemangioma versus dermoid cyst; however, other etiologies such as orbital lymphoma or rhabdomyosarcoma couldn’t be ruled out.
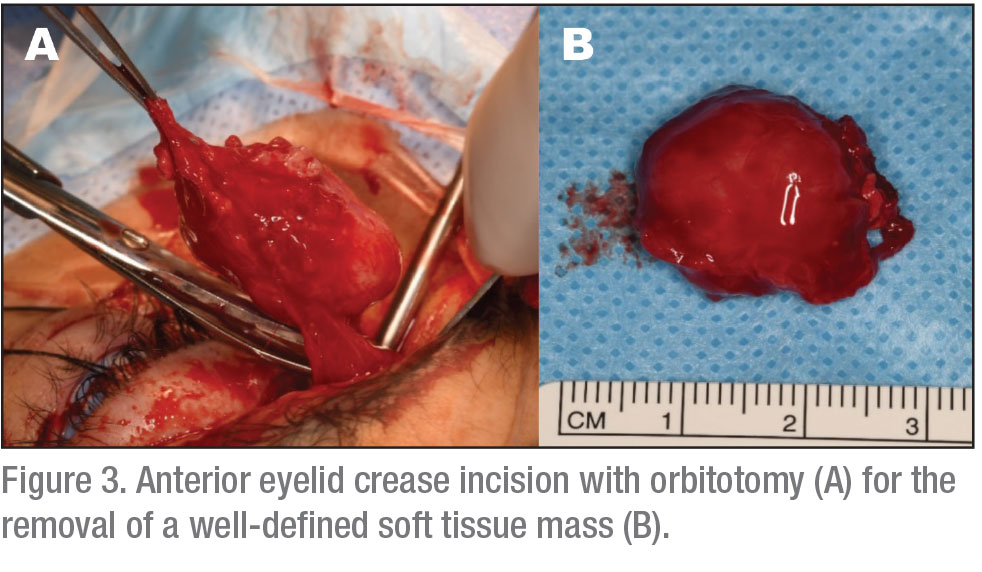
The patient underwent left orbitotomy through an upper eyelid crease incision, with complete removal of the tumor followed by intraorbital injection of sub-Tenon’s kenalog (See Figure 3). The tumor was noted to be adherent to surrounding structures.
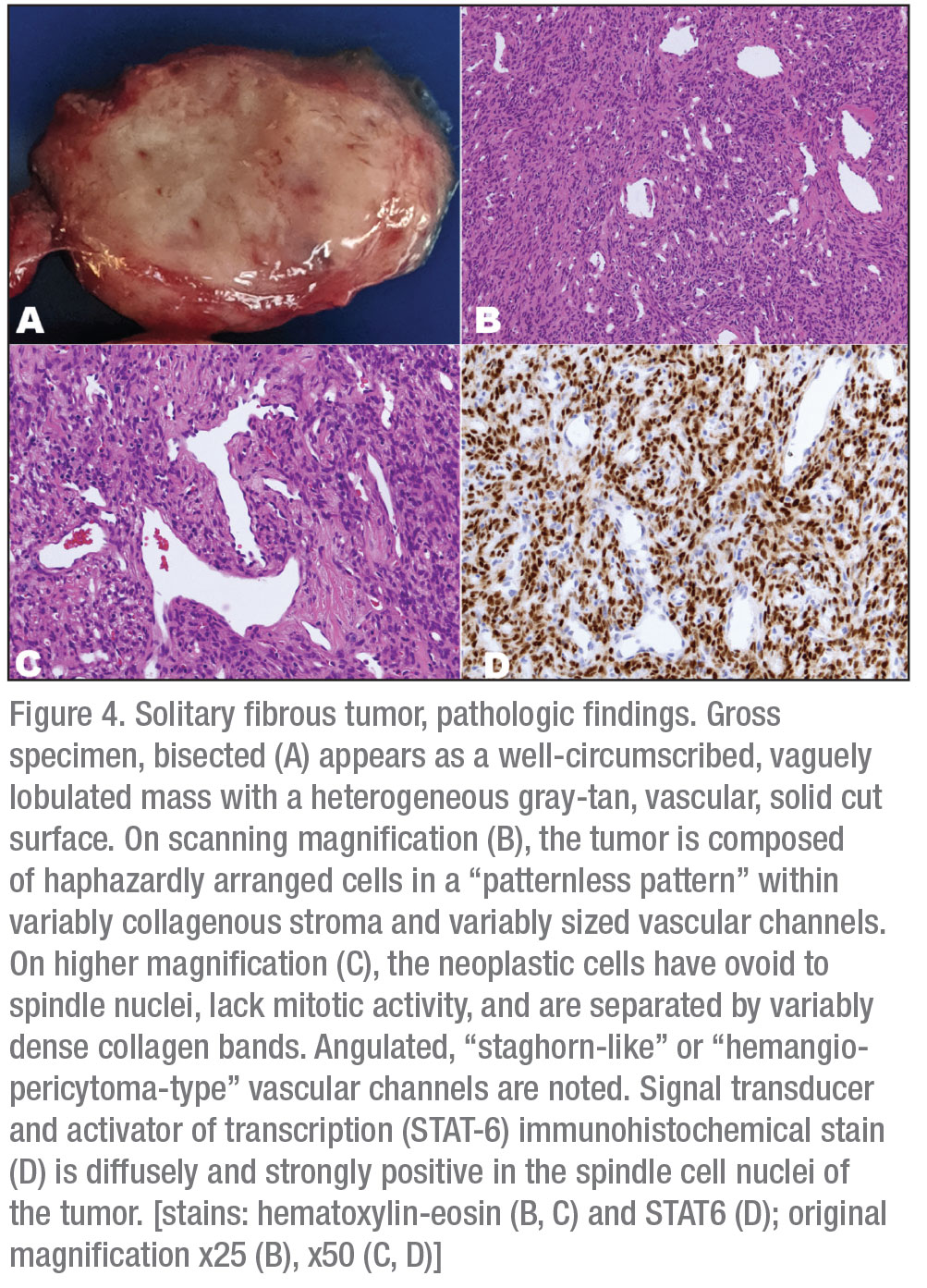
Microscopic examination (Figure 4) showed a well-circumscribed mass composed of cells with spindle nuclei in a variably cellular arrangement, without nuclear overlapping, with intervening foci of keloidal coarse collagen. Prominent vascular channels—both small and capillary-sized and large, branching, hyalinized (hemangiopericytoma-type)—were present throughout the lesion. Scattered lymphocytes and occasional lymphoid aggregates were also noted. Immunohistochemical stains showed that the neoplastic cells express diffusely STAT-6 (nuclear staining), consistent with solitary fibrous tumor. There was no evidence of appreciable nuclear atypia, mitotic figures or necrosis. The Ki-67 proliferative index was 1-2 percent.
Discussion
 |
A wide range of lesions can occur in the orbit, and the differential diagnosis in children varies from that in adults. Lesions can be classified by tumor type; they include cystic, vascular, neural (optic nerve, peripheral nerve, meningeal), inflammatory, osseous and fibrocystic, myogenic, metastatic, lacrimal gland, lymphoid, adipose and histiocytic tumors. The most common orbital tumors in children are cystic lesions, and the most common malignancy in children is rhabdomyosarcoma.1 Understanding the relative incidence of different orbital tumors in each age group can help elucidate the diagnosis when used in conjunction with the patient’s clinical history and radiographic findings.
The most common orbital cystic lesion in children is the dermoid cyst. This lesion typically forms along orbital suture lines from epithelial cells that become trapped during embryogenesis. The most common site is near the superotemporal orbital rim near the zygomatic-frontal suture site.2,3 CT imaging typically reveals an extraconal, well-defined mass with an enhancing wall and non-enhancing lumen. However, some cases may show denser areas which correspond to the accumulation of keratin and other epithelial debris within the lumen of the cyst. MRI shows a well-defined, round to ovoid structure with variable signal intensity based on the material in the lumen, and typically no enhancement with contrast.4 Histopathology reveals the cyst lined by surface epithelium.2 A small orbital dermoid cyst can be observed if asymptomatic; however, in most cases, the cyst gradually enlarges and can rupture or form a cutaneous fistula, necessitating treatment. Treatment involves complete excision of the cyst, being careful to remove the lesion with the capsule intact. If the cyst ruptures, irrigation and removal of the cyst remnants should be performed to minimize risk of recurrence or chronic granulomatous inflammation.2-4
Vascular lesions include tumors such as capillary hemangioma and lymphangioma. Capillary hemangioma is the most common vascular tumor of childhood.1 This lesion increases in size for the first one to two years, then stabilizes and gradually regresses.2,3 They have variable appearance on CT and MRI, ranging from round or ovoid circumscribed masses to irregular or diffuse lesions that enhance with contrast.4 Microscopic exam often shows lobules of vascular channels and proliferating endothelial cells separated by connective tissue septa.2 Management is aimed at preventing and treating amblyopia and strabismus. If vision isn’t threatened, the lesion can be monitored closely. Refraction and occlusive patching should be implemented in patients with amblyopia, and treatment to accelerate regression of the tumor may be considered.
Treatment options include corticosteroids, interferon, radiation, resection and propranolol.2-4 Propranolol therapy can be used orally or topically and has been shown to be an effective and relatively safe option for treating orbital and eyelid capillary hemangiomas.5 Oral or local injection of corticosteroids may also be used, however, but this is associated with risks such as central retinal artery obstruction, eyelid depigmentation, eyelid necrosis and adrenal gland suppression.2 Patients with concomitant cutaneous vascular lesions warrant systemic workup for associated conditions such as Kassabach Merritt and PHACE (Posterior fossa brain malformations, Hemangiomas of the face, Arterial anomalies, Cardiac anomalies, and Eye abnormalities) syndromes.2
Unlike capillary hemangioma, which usually presents in infancy, lymphangioma is a slow-growing lesion that often goes unnoticed until later childhood or teenage years when spontaneous or traumatic hemorrhage may occur and cause sudden, progressive proptosis and/or periorbital soft-tissue swelling.2,3 These lesions typically appear as cystic masses with hemorrhage and varying degrees of enhancement on CT; on MRI, they appear isointense on T1-weighted and hyperintense on T2-weighted images.4 Histopathology shows irregular dilated vascular channels with delicate connective tissue septa.2 Asymptomatic lesions that aren’t sight-threatening are typically observed, since surgical removal is challenging and associated with a high risk of morbidity and recurrence. Vision-threatening lesions (e.g., causing compressive optic neuropathy) and cosmetically unacceptable lesions may be treated with aspiration, sclerotherapy or surgical removal.2-4,6 Exploratory orbitotomy and biopsy is warranted for lesions that can’t be distinguished from rhabdomyosarcoma.
Rhabdomyosarcoma is the most common orbital malignancy of childhood. Patients typically present with proptosis, globe displacement, blepharoptosis, eyelid/conjunctival swelling and in some instances pain, and the mean age of onset is 7 years.1,4
Rhabdomyosarcoma appear hyperdense on CT, isointense on T1- and hyperintense on T2-weighted MRI scans, and they enhance with contrast. They usually present as solid lesions, but can demonstrate cavitary changes that make it difficult to differentiate them from vascular orbital tumors.4 Erosion of surrounding bone may be evident on imaging, reflecting the aggressive nature of the neoplasm. Biopsy shows spindle cells with hyperchromatic nuclei and varying features that delineate the lesions into four types: alveolar; botryoid; embryonal; and pleomorphic. The embryonal type is most common, while the alveolar type is the most malignant.4 Prompt management with biopsy to confirm the diagnosis is key, followed by systemic evaluation for metastasis, and chemotherapy and radiation.
Solitary fibrous tumor (SFT) is a rare fibroblastic spindle cell neoplasm that can develop anywhere in the body, including the orbit. It’s most commonly seen in middle-aged adults; however, several cases have been reported in children.1,7-11 Patients typically present with painless, progressive proptosis or eyelid swelling, and imaging reveals well-circumscribed, variably vascular contrast-enhancing lesions that generally don’t produce bony changes.2-4 On CT, SFT appears isodense to the cerebral cortex and extraocular muscles. MRI shows lesions that are isointense on T1- and isointense to hyperintense on T2-weighted images.4 While helpful in narrowing the diagnosis, these findings aren’t specific for SFT, and it can be difficult to distinguish SFT from other solid orbital lesions such as cavernous hemangioma.
Complete excision is recommended, given the unpredictable nature of this lesion and the potential for malignant transformation.2-4,12 The protein STAT6 is detectable by immunohistochemistry and has been shown to be a highly sensitive and specific diagnostic marker for SFT.13-16 Patients should be followed at regular intervals with postoperative imaging, given the potential for asymptomatic recurrence.
Previously, SFTs were considered separate entities from other fibroblastic mesenchymal tumors such as hemangiopericytomas (HPC). Further histologic review, however, called this classification into question. In 2011, researchers reviewed 41 fibroblastic orbital tumors from the Ophthalmic Registry at the Armed Forces Institute of Pathology and demonstrated that SFTs shared similar morphologic and immunohistochemical features with hemangiopericytomas, giant cell angiofibromas and fibrous histiocytomas.17 Several years later, a gene fusion of NGFI-A binding protein 2 (NAB2) and signal transducer and activator of transcription 6 (STAT6) was identified as the pathogenic mutation underlying SFT. In normal cells, NAB2 functions as a transcriptional repressor in the early growth response (EGR) pathways, which play a critical role in cell growth and proliferation. In SFT cells, the repressor domain in NAB2 is replaced by the activator domain of STAT6, resulting in constitutive expression of the EGR pathway and uncontrolled cell proliferation and growth.18
Subsequent studies have demonstrated the utility of STAT6 as a reliable diagnostic marker for SFT, with sensitivity and specificity of 96 to 100 percent.16,19-20 Additional studies reassessed the clinicopathologic behavior of SFT, given recent advancements in molecular testing. One study was a retrospective review of head and neck lesions previously diagnosed as SFT, HPC, giant cell angiofibroma or orbital fibrous histiocytoma. Using STAT6 as an immunohistochemical marker, researchers found that a greater proportion of head and neck SFT arose in the orbit than previously reported (specifically, 25 percent [22 of 88] of the cases included in the study), and was the second most common site after the sinonasal tract (30 percent). They also reported a higher rate of local recurrence (median time to recurrence: 19 months).21 Another study focused on orbital SFT and found six of 21 cases (29 percent) with a local recurrence, with a median time to recurrence of 4.9 months. Of the six cases with local recurrence, four were asymptomatic and detected via surveillance imaging. All available tissue stained positively for both CD34 (n=21) and STAT6 (n=20).13 These studies highlight the importance of long-term follow-up and surveillance imaging to monitor for local recurrence of SFT.
In summary, a variety of lesions can occur in the orbit in children, with the most common being the dermoid cyst. Orbital SFT is a rare fibroblastic mesenchymal tumor that’s typically seen in middle-aged adults, but can occur in children and should be included in the differential diagnosis of orbital lesions in children, as demonstrated by our case. SFTs are typically benign but have the propensity for local recurrence and metastasis. Thus, close follow-up with surveillance imaging should be performed. REVIEW
1. Shields JA, Shields CL, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: The 2002 Montgomery Lecture, part 1. Ophthalmology 2004;111:5:997-1008.
2. Shields JA, Shields CL. Atlas of Orbital Tumors. Philadelphia: Lippincott Williams & Wilkins, 1999.
3. Nicholson DH, Green WR. Pediatric Ocular Tumors. New York: Masson Publishing USA, 1981.
4. Shields JA, Shields CL. Eyelid, Conjunctival, and Orbital Tumors: An Atlas and Textbook. 3rd ed. Philadelphia: Wolters Kluwer, 2016.
5. Vohra V, Gupta P, Malik PK, Pathak A. Propranolol therapy in a case of capillary hemangioma. Oman J Ophthalmol 2015;8:3:191–193.
6. Schwarcz RM, Ben Simon GJ, Cook T, Goldberg RA. Sclerosing therapy as first line treatment for low flow vascular lesions of the orbit. Am J Ophthalmol 2006;141:2:333-9.
7. Vu, A.F., et al., Recurrent orbital solitary fibrous tumor in a 12-Year-Old. Ocul Oncol Pathol 2017;3:2:83-86.
8. Alexandrakis G, Johnson TE. Recurrent orbital solitary fibrous tumor in a 14-year-old girl. Am J Ophthalmol 2000;130:3:373-376.
9. Lucci LM, Anderson RL, Harrie RP, et al. Solitary fibrous tumor of the orbit in a child. Ophthalmic Plast Reconstr Surg 2001;17:5:369-373.
10. Demirci H, Shields CL, Eagle RC Jr, Shields JA. Giant cell angiofibroma, a variant of solitary fibrous tumor, of the orbit in a 16-year-old girl. Ophthalmic Plast Reconstr Surg 2009;25:5:402.
11. Blandamura S, Alaggio R, Bettini G, et al. Four cases of solitary fibrous tumour of the eye and orbit: One with sarcomatous transformation after radiotherapy and one in a 5-year-old child’s eyelid. J Clin Pathol 2014;67:3:263-267.
12. Sagiv O, Bell D, Guo Y, et al. Pathological features and clinical course in patients with recurrent or malignant orbital solitary fibrous tumor/hemangiopericytoma. Ophthalmic Plast Reconstr Surg 2019;35:2:148-154.
13. Blessing NW, Bermudez-Magner JA, Fernandez MP, et al. Solitary fibrous tumor of the orbit: A case series with clinicopathologic correlation and evaluation of STAT6 as a diagnostic marker. Ophthalmic Plast Reconstr Surg 2020;36:2:164-71.
14. Cheah AL, Billings SD, Goldblum JR, et al. STAT6 rabbit monoclonal antibody is a robust diagnostic tool for the distinction of solitary fibrous tumour from its mimics. Pathology 2014;46:5:389.
15. Yoshida A, Tsuta K, Ohno M, et al. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am J Surg Pathol 2014;38:4:552-559.
16. Doyle LA, Vivero M, Fletcher CD, et al. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol 2014;27:3:390-395.
17. Furusato E, Valenzuela IA, Fanburg-Smith JC, et al. Orbital solitary fibrous tumor: encompassing terminology for hemangiopericytoma, giant cell angiofibroma, and fibrous histiocytoma of the orbit: Reappraisal of 41 cases. Hum Pathol 2011;42:1:120-128.
18. Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 2013;45:2:180-185.
19. Cheah AL, Billings SD, Goldblum JR, et al. STAT6 rabbit monoclonal antibody is a robust diagnostic tool for the distinction of solitary fibrous tumour from its mimics. Pathology 2014;46:5:389.
20. Yoshida A, Tsuta K, Ohno M, et al. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am J Surg Pathol 2014;38:4:552-559.
21. Smith SC, Gooding WE, Elkins M, et al. Solitary fibrous tumors of the head and neck: A multi-institutional clinicopathologic study. Am J Surg Pathol 2017;41:12:1642-1656.


